why do the sinoatrial nodal cells set heart rate?
set frequency of action potentials of heart muscle b/c they lack a stable membrane potential between successive action potentials
display progressive slow depolarization beteen action potentials
what determines the primary pacemaker?
rate of phase 4 depolarization
fastest -> slowest: sinoatrial node, atrioventricular node, purkinje fibers
why is there a delay between atrial & ventricular contractions?
slower conduction velocity at the atrioventricular node
0.05 m/s compared to 1m/s thru the atria (coupled via gap junctions)
**functionally important for complete filling of ventricles to occur before contraction**
describe the electrical pathway in the heart
- action potential generated at sinoatrial node
- traverses right atrium & passes to left atrium via Bachmann’s bundle
- atrioventricular node
- bundle of His
- R + L bundle branches to apex
- up walls of ventricles via Purkinje fibers
Nernst potential for sodium
+60mV
Nernst potential for potassium
-90mV
Nernst potential for calcium
+130mV
result of fast voltage gated Na+ channels in cardiac tissue
activated at -70mV
rapid opening via m gate & rapid closure via h gate
inward membrane current
depolarizes cell (increase of membrane potential/interior becomes more positive)
Kir 2.1
aka inward rectifier
opens at negative voltages
sets stable negatie reting membrane potential of atrial & ventricular muscles cells
close as membrane potential becomes more positive
Ito
aka transient outward K+ current
opens rapidly upon depolarization of membrane
closes rapidly generating transient repolarizing force in ventricular & atrial muscle
IKr & IKs
aka rapid & slow components of delayed rectifier K+ current
closed at negative voltages
open when membrane potential becomes more +
mainly responsible for repolarization of action potentials in heart
difference between IKr & IKs
IKr has faster inactivation rate than IKs
IKr current is larger→ more important current in facilitating repolarization of action potentials
T-type calcium ion channels
Tiny conductance & Transient openings
predominantly in pacemaker & atrial tissue
open at -55mV
inactivate fairly rapidly
relatively small conductance compared to L-type
L-type calcium ion channels
Large conductance & Long Lasting openings
found throughout heart
open @ -40mV
inactivate more slowly comapred to T-type
cardiac ion channels
- Na+: fast voltage gated Na+ channels
- inward flow→ depolarizes/increases potential
- K+: Kir 2.1, Ito, IKr, IKs
- outward flow→ repolarizes/decreases potential
- Ca2+: T-type & L-type (predominant)
- inward flow→ depolarizes/increases potential
If
aka Funny current
mixed conductance channel
permeable to Na+ & K+
activated by hyperpolarization (@ - membrane potentials) & cAMP (ligand gated)
small conductance channel
@-60mV, driving force of Na+ influx greater than K+efflux
importance of NCX
aka sodium calcium exchange antiporter
3 Na+ influx w/1 Ca2+ out
generates inward membrane current by charge difference (electrogenic)
removes Ca2+ during muscle relaxation
what ion channels are not present at the sinoatrial node & the effect?
Kir 2.1 (inward rectifier K+ channels)
no stable resting membrane potential during phase 4
maximum diastolic potential
most negative membrane potential observed in SA or AV nodal cells
Phase 0 of nodal action potential
aka upstroke
produced by opening of voltage-dependent L-type Ca2+ channels
causes influx of Ca2+
threshold of -40mV
5V/s
Phase 3 of nodal action potential
aka repolarization
produced by closure of L-type Ca2+ channels & opening of delayed rectifier IKr & IKs
stops influx of Ca2+ & efflux of K+
Phase 4 of nodal action potential
aka pacemaker potential
membrane potential more negative→ IKr & IKs close & funny channels open→ stop movement of K+ & influx of Na+
membrane potenial increases
@ -55mV T-type Ca2+ channels open→continues depolarization
@ -40mV L-type Ca2+ channels open starting phase 0
Phase 0 of ventricular action potential
aka upstroke
200V/s
opening of fast voltage-dependent Na+ channels→ influx of Na+→ depolarization
as membrane potential becomes more +, Kir 2.1 channels close→ membrane less permeable to K+
phase 1 of ventricular action potential
aka early repolarization
produced bu closure of fast voltage-gated Na+ channels
opening of transient outward voltage-gated K+ channels (Ito)
phase 2 of ventricular action potential
aka plateau
opening of voltage-dependent slow L-type Ca2+ channels→ influx of Ca2+
downward slope produced by opening of slow delayed rectifier K+ channels (IKs)→ efflux of K+
NCX antiport also contributes maintaining the plateau
Phase 3 of ventricular action potential
aka rapid repolarization
produced by closure of L-type Ca2+ channels & opening of several K+ channels
IKs & IKr open until reaching -60mV
Kir 2.1 complete final phase of repolarization
phase 4 of ventricular action potential
aka resting membrane potential
IKs & IKr are closed
Kir 2.1 (inward rectifier) remains open & maintains resting membrane potential
why are epicardial action potentials shorter in duration than endocardial action potentials?
greater expression of Ito (transient outward K+ channels)→ creates phase 1 notch
depolarization spreads endo to epi & repolarization spreads epi to endo→ thus epi must be shorter→ stops adjacent cells from reactivating those surrounding them
intrinsic firing frequencies of pacemaker cells
sinoatrial cells= 60-100 bpm
atrioventricular cells= 40-60 bpm
Purkinje cells= 20-40 bpm
overdrive-suppression
rapid firing of sinoatrial node causes secondary (AV node) & tertiary (Purkinje) pacemakers to fire at SA node rate (faster than their intrinsic rates)
what current(s) contribute to the action potential in both SA node & ventricular cells?
L-type Ca2+ current
calsequestrin
protein that binds Ca2+ in SR
closely associated w/SR terminal cisternae
why are T-tubules wider in cardiac tissue than skeletal?
to reduce the prospect of Ca2+ ion depletion
where are L-type Ca2+ channels located?
aka DHPR (dihydropyridine receptors) & CaV 1.2
sarcolemma along T-tubule
about 30% of Ca2+ for each contraction enters this way
methods for restoration of Ca2+ after cardiac muscle contraction
- reuptake by sarcoplasmic reticulum via SERCA2 pump (70%)
- removed from cell by NCX exchanger (28%)
- removed from cell by plasmalemmal Ca2+ ATPase (2%)
describe mechanism of Ca2+ reuptake by the sarcoplasmic reticulum
SERCA2 (sarcoplasmic endoplasmic reticulum calcium ATPase) regulated by phospholamban
phospholamban dissociates from SERCA2 when phosphorylated at ser16 by PKA & thr17 by CaM kinase II
phospholamban dissociation increases rate of Ca uptake
similarities between skeletal & cardiac muscle
- striated
- interdigitating thin & thick filaments displaying A & I bands
- thin filament regulatory proteins: tropomysosin & troponin
- cross bridge cycle is identical
differences between cardiac & skeletal muscle
- wider T-tubules in cardiac muscle
- T-tubules enter at Z-lines in cadiac muscle
- mechanism of Ca2+ release from sarcoplasmic reticulum
- fewer DHPRs in cardiac muscle
- Ca2+ doesn’t enter skeletal muscle & thus doesn’t need to be removed
- Skeletal DHPR= CaV 1.1/Cardiac DHPR=CaV 1.2
- Skeletal RyR1/Cardiac RyR2
- no physical connection between DHPR & RyR in cardiac
*
- no physical connection between DHPR & RyR in cardiac
sites of of ATP usage in cardiac muscle
- bind S1 myosin head for cross bridge cycle
- SERCA2
- PMCA: remove Ca2+ from cell
- Na+/K+ ATPase
- adenylate cyclase (AC)
what are the primary metabolites utilized by the normal adult heart?
free fatty acids (60-70%) & carbohydrates
what is the advantage(s) of the long plateau observed in ventricular muscle action potentials?
for every contraction period (systole), there is a resting period (diastole) during which the heart can refill w/blood
why can’t cardiac muscle remain in a state of sustained (tetanic) contraction?
b/c cardiac muscle AP duration is musch longer→ longer refractory period
fast sodium current inactivates rapidly, but cannot be reactivated until negative membrane potentials are reached (-70mV) during repolarization
optimal length
Lo
narrow range of whole-muscle stretched lengthin which active tension is maximal
diastole
cardiac muscle cells are relaxed
cytoplsmic Ca2+ is very low
blood enters ventricles
how is passive tension generated in ventricular muscle?
blood entering the ventricles stretches the cells to increase volume
titin is grestest contributor (same as in skeletal muscle)
preload
aka end diastolic volume
volume in the vnetricle at the end of the diastolic phase
what is the difference in normal working range between skeletal & cardiac muscle?
skeletal= total tension ranges around optimal length
cardiac= below optimal length
Frank-Starling Law
stroke volume of the heart increases in response to an increase in the volume of blood filling the heart (the end diastolic volume) when all other factors remain constant
afterload
in left ventricle= aortic diastolic pressure
load the muscle must overcome to open the aortic valve & eject blood
when ventricular pressure exceeds afterload→ aortic valve opens
what is the only limiting factor of Vmax of ventricular shortening velocity?
myosin ATPase
what happens to shortening velocity for a certain afterload as preload increases?
shortening load will increase
blood ejected faster b/c force of contraction is increased
how can Vmax (velocity of shortening) be increased?
increased inotropic status
ex. increase in sympathetic stuimulation
Fmax
theoretical afterload (aortic pressure) so high that muscle cannot contract
how can Fmax be increased?
increased inotropic status
ex. increase in sympathetic stimulation
effects of increased preload
- increased velocity of shortening at a given afterload
- increased Fmax w/o changing Vmax
effects of increasing inotropy
- increases velocity of shortening at a given afterload
- increases Fmax & Vmax
how is an electrocardiogram recorded?
epicardial cells are + compared to endocardial cells when heart is at rest
during AP: epicardial cells change from + to - → this change is recorded by ECG
conducted change is recorded via extracellular fluid to the surface of the skin
electrodes detect movement or spread of electrical activity throughout the heart
recorded signal is much smaller than reality
what causes a positive deflection to be recorded?
depolarization (action potential) toward the positive electrode (cathode)
OR
repolarization traveling away form positive electrode
what is recorded when the whole tissue is depolarized?
a flat line at the isoelectric point
b/c there is no potential difference to measure between two points
what causes a negative deflection to be recorded?
depolarization (action potential) travelling away from positive electrode
OR
repolarization travelling toward positive electrode
Einthoven’s triangle
arrangement of standard limb leads (I, II, & III)
records electrical activity in frontal plane through heart
Dr Einthoven invented first practical ECG in 1903
Lead I
+ electrode on LA
- electrode on RA
0º horizontal plane; bipolar lead
Lead II
+ electrode on LL
- electrode on RA
Lead III
+ electrode on LL
- electrode on LA
what physiological event corresponds to the P wave?
atrial depolarization→ contraction of atria
depolarization as AP spreads away from SA node throughout atria @ 1m/s
speed due to gap junction connectivity througout atria
1st 1/3 is RA depolarization, 2nd 1/3 is RA & LA depolarization, & 3rd 1/3 is LA depolarization b/c RA depolarizes 1st
what physiological event corresponds to the QRS complex?
ventricular depolarization→ventricular contraction
depolarization (AP) spreads down bundle branches to apex @ 5m/s
results in positive deflection= R wave
normal 0.08-0.10seconds
atrial repolarization is also occuring, but obscured by signal from ventricles
what physiological event corresponds to the ST segment?
period of time that the ventricles are fully depolarized
duration of plateau phase of ventricular action potential
last about 120sec
elevated in MI
depressed when heart is hypoxic (coronary insufficiency)
what physiological event corresponds to the T wave?
ventricular repolarization
what physiological event corresponds to the QT interval?
period from beginning of ventricular depolarization to end of ventricular repolarization
ventricular systole takes place
normal <400ms (0.4 seconds)
what physiological event corresponds to the PR segment?
AP slowly (0.05m/s) transmitted thru AV node
electrically neutral period
what is the most important factor to analyze about segments?
change from the isoelectric line
what is the most important factor to analyze about intervals?
duration
what physiological event corresponds to the PQ/PR interval?
P wave & PR segment
time between atrial excitation & beginning of ventricular excitation
normal is 0.12-0.20
shortens w/increased HR & lengthens w/decreased HR
>0.20 seconds→AV conductions block (AV node of bundle of His)
what physiological event corresponds to the Q deflection?
spread of the AP across the interventricular septum from L to R
seen on lead II, but not all leads
what physiological event occurs R→S?
downward deflection created by depolarization (AP) moving away from positive electrode (apex)
dominated by larger L ventricle
what physiological event corresponds to the T wave?
results from ventricular repolarization
base 1st/apex last
+ deflection created by dominant repolarization moving away form + electrode
typically lasts 160ms
broader w/lower amplitude b/c repolarization is slower than depolarization & less unidirectional
segment v. interval
segments are on isoelectric line
intervals include a waveform
how does coronary insufficiency present on an ECG?
ST segment depression
what is a subendocardial infarct?
infarct does not affect teh full thickness of L ventricular wall
what can cause a prolonged QT interval?
myocardial damage
coronary ischema
conduction abnormalities: genetic syndromes including long QT
Long QT
many causes
predisposes to ventricular arrhythmias
delays repolarization phase in ventricualr tissue
cause of LQTS1
loss of function of IKs
cause of LQTS2
loss of function IKr
cause of LQTS3
gain of function to INa
faster upstroke of ventricular action potential
what physiological event corresponds to the TP segment?
heart totally quiescent
diastole
heart refills w/blood
which wire plays no role in the ECG waveforms?
RL
it’s neutral lead
exisits to complete the electrical circuit only
how are augmented limb leads created?
electrical signal from 2 limb electrodes is simultaneously fed into negative terminal
produces and average signal located roughly in the middle of the chest
aVL
augmented Vector Left
+LA
- RA & -LL
- 30º
aVR
augmented Vector Right
+RA
- LA & -LL
- 150º
aVF
augment Vector (left) Foot
+LL
-RA & -LA
+90º
precordial leads
chest leads
all +
share common negative= Wilson’s central terminal
record electrical activity in transverse plane thru heart
progression from - QRS @ V1 to + QRS @ V6
time segments on ECG trace paper
on horizontal axis
small block= 0.04 sec
large block= 0.2 sec
voltage segments on ECG trace paper
on vertical axis
small block= 0.1mV
large block= 0.5mV
what does the height of QRS complex tell us?
greater height of QRS complex→ more aligned spread of depolarization on a particular vector
if spread of depolarization is directly towards a lead, then QRS will be most + on that lead
exact calculation of heart rate from ECG trace
(# of small blocks * 0.04sec)= (x seconds/beat)
(60 seconds/minute) / (x seconds/beat)= bpm
quick estimate of HR by ECG trace paper
- 1 large block= 300
- 2 large block= 150
- 3 large block= 100
- 4 large block= 75
- 5 large block= 60
- 6 large block= 50
- 7 large block= 43
- 8 large block= 37
normal PR interval
0.12-0.20 seconds
normal QRS complex duration
0.10 seconds
normal QT interval duration
<1/2 RR interval at rest
normal sinus rhythm
1:1:1 ratio P wave: QRS complex: T wave
- rhythm: regular
- rate: 60-99bpm
- QRS duration: normal (<0.10seconds [3 small blocks])
- P wave: visible before each QRS
- PR interval: normal (0.12-0.20 seconds/<5small blocks)
junctional rhythm
SA node is not functional→ AV node is primary pacemaker→ inverted or no P waves
- rhythm: regular
- rate: 40-60bpm
- QRS duration: normal
- P wave: inverted in lead II or not visible
sinus tachycardia
- rhythm: regular
- rate: >100bpm
- QRS duration: normal (<0.10seconds [3 small blocks])
- P wave: visible before each QRS
- PR interval: normal (0.12-0.20 seconds/<5small blocks)
causes: (sympathetic innervation), exercise, fever
sinus bradycardia
- rhythm: regular
- rate: <60bpm
- QRS duration: normal (<0.10seconds [3 small blocks])
- P wave: visible before each QRS
- PR interval: normal (0.12-0.20 seconds/<5small blocks)
causes: (parasympathetic innervation), highly trained athletes, pts on beta blockers
Long QT syndrome ECG
normal QT interval duration= 400ms
QTc (men)= 450ms/QTc (women)= 470ms
corrected by Bazzett’s formula: QTc= QT duration/ square root(RR interval [HR])
1º AV block
first degree atrioventricular conduction block
partial block of AV node→ slower conduction thru AV node
- rhythm: regular
- rate: 60-99bpm
- QRS duration: normal (<0.10seconds [3 small blocks])
- P wave: visible before each QRS
- PR interval: >0.20 seconds/>5small blocks)
causes: fibrosis, inferior MI, OR induced by enhanced vagal tone, athletic training, beta blockers, Ca2+channel blockers (can be normal finding)
tx: none needed, but may increase risk of more serious heart block types
Wenkebach’s heart block
Mobitz type I 2º heart block
pregressive lengthening of PR interval w/each beat until P wave fails to result in QRS complex then resets
cause: conduction block in AV node
tx: usually benign→ seen in children, athletes, pt w/elevated vagal tone
2º heart block
PR interval: > 0.25 seconds (>6 small blocks)
sometimes, not always conduction thru AV node fails→ P wave occurs, but does not produce QRS complex
intermittent conduction failure w/subsequent loss of ventricular contraction
Mobitz type II 2º heart block
no lengthening of PR interval
sudden, unexpected loss of AV conduction & ventricular activation
wide QRS b/c excitation is spreading cell-to-cell instead of thru the His-Purkinje system
more dangerous than Mobitz type I
could lead to cardiac arrest
tx: pacemaker implant
3º heart block
complete failure of conduction from atria to ventricles
no relation between P waves & ventricular contraction
- rhythm: regular
- rate: atria @ 60-99bpm/ventricles @ 20-40bpm
- PP interval short than RR interval
- QRS duration: broad QRS b/c excitation is spreading cell-to-cell instead of thru the His-Purkinje system
- P wave: 2-4 for each QRS complex
- PR interval: normal (0.12-0.20 seconds/<5small blocks)
Atrial Flutter
atrial focus activate atria NOT SA node
- rhythm: regular
- rate: usually>100bpm up to 300bpm
- QRS duration: normal (<0.10seconds [3 small blocks])
- P wave: repetitive, obscuring baseline
- “saw tooth” appearance
- 2-4/QRS complex
Atrial Fibrillation
lack of coordination between atria & ventricles
re-entry current occurring in atria
- rhythm: irregularly irregular
- P wave: none distinguishable
not directly fatal, but blood can pool in poorly mixed areas→ thrombus→ embolism
re-entry current
damaged/diseased cardiac tissue conducts so slowly that AP excites adjacent repolarized tissue
re-entry of AP
result: non-rhythmic cycyles of contraction & relaxation
Ventricular Fibrillation
re-entry current in ventricles
TOTAL absence of any pattern
arrythmic; no R waves to determine HR
result: decrease stroke volume to fatally low level
where does a pulmonary embolism originate?
as a R atrium thrombus
what can occur if a thrombus is present in the left atria?
coronary or cerebral embolism→ MI or stroke
Monomorphic Ventricular Tachycardia
caused by irritable ventricular focus→ discharges premature impulses for 3+ beats w/o interruption
- rhythm: regularly irregular
- QRS duration: broader than normal b/c impulses are conducted cell-to-cell instead of His-Purkinje system
- all look the same
- no distinguishable P or T waves
Polymorphic Ventricular Tachycardia
beat-to-beat variation in QRS morphology
each ventricular impulse can be generated form a different place
QRS may be taller or wider
common type: Torsades de pointes
expect consultation needed
effects of ventricular tachycardia
decrease cardiac output→ hypotension, collapse, acute heart failure, & possible myocardial perfusion w/degeneration to ventricular failure
caused by extreme HRs & uncoordinated atrial contracrion
decreased cardiac output may also
PVCs
premature ventricular contractions originating in ventricles from foci w/in His-Purkinje system
- rhythm: dependent on uni or multifocal
- rate: any
- QRS duration: broad, strangely shaped
- P wave: absent
- PR interval: normal (0.12-0.20 seconds/<5small blocks)
- T wave: - if net + QRS, or vice versa
quadrigeminy
3:1 PVC
couplet
2 PVC together surrounded by normal sinus rhythm
bigeminy
1:1 normal beat to PVC
trigeminy
2:1 normal to PVC beats
Wolff-Parkinson-White Syndrome
type of pre-excitation syndrome w/delta waves
consequence of abnormal conduction via accessory pathway across annulus fibrosus cordis (often Bundle of Kent)→ can conduct antergrade (rare), retrograde (15%), or both (in majority)
- rhythm: regularly irregular
- QRS duration: wide; >120ms
- delta wave= slurred onset
- P wave: visible before each QRS
- PR interval: short; 0.<12 seconds; effectively absent
- secondary ST or T wave changes
LBBB
aka left bundle branch block
broad QRS
depolarization spread across septum reversed (R→L)
large - & elongated S waves in V1-V3
nothed or slurred R wave in V6
repolarization endo to epi (reversed)→ inverted T wave in V6
RBBB
aka right bundle branch block
broad QRS
depolarization spread normal across spectum (L→R)
delayed depolarization of RV generated seconds R wave→ seen in V1
slurred Swave in V6
inverted T wave in V1
regular rhythm
RR interval are identical for wach heart beat
rhythm is maintained
regularly irregular rhythm
pattern of beats that repeats
2º heart block
irregularly irregular rhythm
no underlying regularity
inconsistent RR interval (HR)
ex. atrial fibrillation
define mean electrical axis (MEA) of heart & what is the normal range?
net direction of electrical conduction during ventricular depolarization
tells us:
- orientation of heart
- size of ventricular chanbers
- conduction block
normal range= +90 to -30º
what change in MEA does right ventricular hypertrophy cause?
right axis deviation= +90 to +180º
CAN also be normal finding in children & tall thin adults
QRS is - in lead I & + in aVF
what are the cause(s) of right axis deviation?
- normal in children & tall thin adults
- right ventricular hypertrophy
- chronic lung dz w/pulmonary HTN
- pulmonary embolism
what change in MEA does left ventricular hypertrophy cause?
left axis deviation= -30 to -90º
QRS is + in lead I & - in lead II and aVF
what are the cause(s) of left axis deviation?
- left ventricular hypertrophy
- inferior MI
- emphysema→ heart moved posteriorly due to hyperexpanded lung pushing L ventricle (thus MEA) into upper R quadrant
semi-quantitative method for determining MEA
- draw radial axis
- determine net + or - for lead I
- draw 90º arc in either direction from corresponding end of arrow
- repeat for all 6 limb leads
- 30º segment where all 6 lines are present is where the pt’s MEA will be found
Net zero method for determining MEA
**only applicable if one limb lead shows net zero
- determine net zero lead
- go to perpendicular lead
- If QRS of perpendicular lead is net +, then + end is pt’s MEA. If QRS of perpendicular lead is -, then -end is pt’s MEA.
Quick & dirty method for MEA determination
- normal MEA
- Lead I= +
- Lead aVF= +
- Right axis deviation
- Lead I= -
- Lead aVF= +
- left axis deviation
- Lead I= +
- Lead aVF= -
what occurs during phase 3 of the cardiac cycle? and what is the corresponding electrical event on the ECG?
atrial contraction
P wave tha induces atrial contraction occurs just before this phase
what phase of the cardiac cycles does the QRS complex preceed?
ventricular contraction/systole
phase 4-5
what electrical event on an ECG immediately preceeds phases 6-7 (ventricular relaxation/diastole)?
T wave
what event & cardiac phase is associated with the ST segment?
phase 5= rapid phase of ventricular ejection
what physical events occur during systole?
ventricular contraction & ejection
what physical events occur during diastole?
ventricular relaxation & filling phase
what happens when left artial pressure exceeds left ventricular pressure?
typically aroun 10mmHg
mitral valve opens
rapid passive filling of LV→ LV volume increases
aortic pressure is high b/c ventricle has just finished ejection phase & aortic valve closed
what is the approximate volume of left ventricular end systolic volume?
50mL
what phase of the cardiac cycle occur during diastole?
phases 1-3
- 1: rapid ventricular filling
- 2: diastasis (slow ventricular filling)
- 3: atrial contraction
- only contributes 5-10% of ventricular volume
a wave
wave of increased L atrial pressue on atrial pressure curve immediately following P wave on ECG
also causes small rise in ventricular pressure & volume
immediately followed by mitral valve closure
what created the S1 sound?
mitral valve closure
what begins systole?
mitral vavle closure
isovolumetric contraction phase (phase 4)
c wave
positive deflection on atrial pressure curve due to teh bulging of the mitral valve into the atrium as the ventricle begins to contract
what causes the aortic vavle to open?
when left ventricular pressure exceeds aortic pressure
occurs during phase 5 aka rapid ventricular ejection phase
what reduces the rate of blood flow during the slow ventricular ejection (6) phase?
reduced ventricular pressure
due to reduced # of contracting cardiomyocytes as repolarization begins
what phase is at the beginning of diastole?
isovolumetric relaxations (7) phase
marked also by the closing of the aortic valve that creast the S2 heart sound as it closes
what creates the S2 heart sound?
aortic valve closure
how are heart sounds produced?
the sudden stretching & subsequent vibration of compliant valves, surrounding connective tissue, & vessels creates sound
NOT the closure itself
Windkessel effect
transient increase in aortic pressure as blood starts to flow back into ventricle shutting the aortic valve cusps
turbulent flow helps support coronary circulation & creates dicrotic notch in aortic pressure curve
v wave
positive deflection of left atrial pressure curve created by atrial filling
venous pressure wave
jugular vein pressure waves created by events on R side of heart
- a-wave: RA contraction
- c-wave: tricuspid valve bulging into RA
- x-descent: relaxation of RA
- v-wave: filling of RA against closed tricuspid valve
- y-descent: fall of jugular venous pressure due to rapid phase of ventricular filling
what does point A on a pressure-volume loop coorespond to?
end systolic volume
mitral valve opens
what does Phase I on a pressure-volume loop coorespond to?
rapid ventricular filling
diastasis
atrial contraction
what does Point B on a pressure-volume loop coorespond to?
end diastolic volume (preload)
beginning of systole
mitral valve closes (S1 sound)
Preload
left ventricular wall stress at end of diastole
normally related to volume in LV at end of diastole
what does Phase II on a pressure-volume loop coorespond to?
isovolumteric contraction of left ventricle
what does Point C on a pressure-volume loop coorespond to?
aortic semi-lunar valve opens
estimate of diastolic blood pressure (afterload)
what does Phase III on a pressure-volume loop coorespond to?
rapid phase of ventricular ejection
estimate of systolic BP (highest point of curve- estimate of maximal afterload pressure)
slow phase of ventricular ejection
what does Point D on a pressure-volume loop coorespond to?
aortic valve closes (w/S2 sound)
TRUE end systolic volume
same volume as A in health
marks beginning of ventricular diastole
what does Phase IV on a pressure-volume loop coorespond to?
isovolumetric relaxation
local blood flow
aka intrinsic blood flow
mechanisms by which individual organs control their own blood flow
metabolic regulation of blood flow
arterioles sense their external environment (interstitial fluids) for metabolites & paracrine agents & respond appropriately
cause local dilation or constriction
What are the metabolites that influence arteriolar blood flow?
adenosine
lactate
H+
K+
what are the paracrine agents that influence arteriolar blood flow?
angiotensin II
Histamine (vasodilaton)
bradykinin
prostaglandins (F family→ constriction; E family→ dilation)
How does O2 concentration influence arteriolar blood flow?
decreased O2→ local dilation (to get more)
Does CO2 influence arteriolar blood flow? If so, how?
Yes
increased [CO2] in the interstitium→ local dilation (to carry out waste)
describe the myogenic reflex in relation to blood flow regulation.
- increased blood flow
- excessive stretch of blood vessels
- stretch activated Na+ channels
- depolarization of smooth muscle cells
- L-type Ca2+ channels
- smooth muscle cell contraction
- vasoconstriction
- decreased blood flow
How can NO production be stimulated?
- paracrine agents
- histamine via H1 receptors
- ACh from cholinergic nerves
- Bradykinin
- increased sheer stress on luminal vessel wall (endothelial cells!!)
- elevation of pressure gradient along long axis of vessel
What reaction does NO synthase catalyze?
L-arginine + O2 + NADPH→ citrulline + NO + NADP
How does NO travel from the epithelial cells to vascular smooth muscle?
simple diffusion b/c it is a lipophilic gas
How does increased [cGMP] lead to vasodilation?
- increased cGMP
- activation of protein kinase G (PKG)
- phosphorylation of phospholamban
- dissociation of phospholamban from SERCA2
- SERCA2 increased rate of Ca2+ uptake
- decreased contractility of vascular smooth muscle
- vasodilation
PKG activation leads to inhibition of myosin light chain kinase reduced Ca2+ sensitivity of myofilaments→ relaxation
How do organs maintain a relatively constant blood flow regardless of perfusion pressure?
autoregulation
myogenic theory explains reduced flow in the presence of increased systemic HTN
metabolic theory explains the vasodilaton & increased flow that results after a sudden decline in perfusion pressure
What organs most clearly display autoregulation?
kidney, heart, & brain respectively
what organ(s) do not display autoregulation?
skin & pulmonary circulation
over what blood pressure range do the heart & brain maintain a constant flow?
60-180mmHg
hyperemia
incerased blood flow to different tissues of the body
active hyperemia
increase in blood flow due to an increase in metabolism
- increased metabolism
- increased release of metabolic vasodilators into extracellular fluid
- dilation of arterioles
- decreased resistance created increased blood flow
- O2 & nutrient supply to tissue increases for duration of increased metabolism
functional hyperemia
aka exercise hyperemia
active hyperemia that occurs during exercise
mostly to type I (aerobic) myocytes
blood flow increass to 80-85% of cardiac output to skeletal muscle from normal of 15-20%
reactive hyperemia
increased blood flow & vasodilaton that occurs in reaction to short interruption of blood flow
temporary occlusion→ buildup of metabolic waste→ vasodilation
longer occlusion→ longer period of subsequent reactive hyperemia
what type of activity displays active hyperemia?
phasic exercise
ex. running
what type of activity displays reactive hyperemia?
occlusive muscle activity
ex. weight lifting
what mechanisms are involved in hyperemic reponse to exercise?
adenosine & K+
maybe sympathetic cholinergic fibers in exercise-induced vasodilation
why does sympathetic activation lead to coronary vasodilation?
- stress induced SNS
- HR & SV increased
- SNS alpha-1 adrengenic receptors→ vasoconstriction
- active hyperemia caused by production of adenosine & NO
- vasodilation
when is reactive hyperemia observable in coronary blood flow?
during systole (contraction)
oddly this means that there is decreased flow when it is needed the most
when is left ventricular coronary blood flow highest & why?
during diastole
b/c ventricular muscle is relaxed→ coronary vessels are unobstructed
when is right ventricular coronary blood flow highest?
during systole
right ventricular does NOT contract w/as musch force as L→ right coronary flow is not impeded
coronary artery pressure rises during systole→ increases driving pressure & R coronary blood flow
less reactive hyperemia compared to left coronary arteries
at rest, approximately what percentage of O2 & cardiac output does the brain consume?
O2= 20%
cardiac output= 14%
what anatomical anstomosis helps to maintain cerebral blood flow?
Circle of Willis
created by the union of internal carotid arteries & basilar artery
how does cerebral blood flow increase?
- increased brain activity
- increased metabolic rate
- raised arterial CO2 (hypercapnia)– active hyperemia metabolite
- vasodilation
cardiac input
aka venous return
what is required physiolgically for blood to return to the heart?
pressure gradient between the right atrium (approximately 2mmHg) & central venous pressure (CVP) (approximately 10mmHg)
why does a venous system pressure gradient of 8mmHg generally lead to a flow of 5L/min through the veins?
- 8mmHg is average pressure gradient fro venous return
- venous return= cardiac input
- cardiac input= cardiac output
- average cardiac output= 5L/min
what is the most important determinate of cardiac output?
venous return
what is the most important determinate of venous return?
central venous pressure
what happens to venous return when right atrial pressure increases?
driving pressure from central veins to RA (CVP)
smaller gradient
decrease in venous return
what happens to venous return when right atrial pressure decreases?
driving pressure from central veins to RA increases (increased CVP)
larger gradient
increased venous return
mean systemic filling pressure
when vascular function curve intercepts x-axis
venous return= 0L/min
this pressure is generated as a result of fullness of circulation
what effect does an increase in blood volume have on the vascular function curve?
increase mean systemic filling pressure
shift entire curve to the right
name 3 parameters that will increase venous return.
= increase cardiac output
- increase blood volume
- decrease venous compliance
- aka increase venous tone
- decrease arteriolar resistance
- more blood shifts to venous system
how does increased venous compliance effect the vascular function curve?
increased venous compliance→ more blood held in venous reservoir of veins→ decreased venous return
curve shifts down & to the left
decreased mean systemic filling pressure
how does decreased arteriolar resistance effect the vascular function curve?
decreased arteriolar resistance= decreased tone→ increased venous volume→ increased venous pressure→ larger gradient (from right atrial pressure)→ increased venous return
mean systemic filling pressure stays the same & maximal venous return/cardiac output is increased→ slope is more extreme
how does arteriolar constriction effect arterial & venous pressures?
increase atrial pressure
decrease venous pressure
what is the independent variable on the vascular function curve?
venous return (L/min)
however the graph is always plotted inversely, so it appears on the y axis
what is the dependent variable on the Starling curve?
starling curve= cardiac function curve
cardiac output (L/min) is dependent
right atrial pressure is independent variable
per the Frank-Starling relationship, how does increased right atrial pressure influence cardiac output?
increased right atrial pressure→ increase in end diastolic volume (EDV)→ increased cardiac output
What factor can shift the cardiac function curve up or down?
cardiac contractility
what methods lead to hypereffectiveness of the heart & their effect on the cardiac function curve?
- increase sympathetic stimulation
- increase circulating epinephrine
- inotropic drug
curve shifts up & left
what methods lead to hypoeffectiveness of the heart & their effect on the cardiac fucntion curve?
- decrease sympathetic stimulation
- loss of ventricular tissue (severe MI)
- inotropic drugs
curve moves down & to the right
Starling’s law
heart contracts more forcefully when filled to a greater degree
what effect does increasing preload have on a pressure-volume loop?
- increased preload= increased ventricular filling (EDV)
- assuming fixed contractility
- increaseds velocity of shortening
- increased stroke volume
- pressure-volume loop changes
- point B shifts up & right
- point C moves right & slightly up
- no change of A, D, or Vmax
how does an increase in venous return affect cadiac output?
- increased venous return
- increases left atrial filling
- increases preload= left ventricular filling (EDV)
- increases sarcomere length
- increases force of contraction
- increases left ventricle stroke volume
- increases cardiac output
how does a normal heart correct increased pulmonary pressure?
increased pulmonary pressure→ increase left atrial pressure→ increases left ventricular filling (EDV/preload)→ greater contractility to eject larger blood volume
caused by right heart pumping more blood (more efficiently) than the left
what effect does increasing afterload have on a pressure-volume loop?
aka increase aortic diastolic pressure w/constant contractility
- required greater left ventricular pressure to open aortic semilunar valve, ie more time
- decreases time foor ventricular ejection
- decreases stroke volume
- decreases cardiac output & increases ESV
- pressure-volume loop changes
- points B & C may move slightly right
- point A shifts right
- point D shift up & right along ESPVR line
how is increased afterload presented clinically?
normal or low systolic aortic pressure
decreased diastolic pressure
elevated left atrial pressure (from back up of blood volume)
usually caused by aortic valve stenosis
what effect does increasing contractility have on a pressure-volume loop?
caused by increasing sympathetic innervation of the heart
- increases slope of ESPVR line
- increases ability of cardiac muscle to shorten during contraction
- increases pressure @ any given EDV
- decrease in ESV
- increases stroke volume
- pressure-volume loop changes:
- point B & C stay the same
- point A shifts left
- point D shifts left & on new ESPVR line
- increase in Vmax
what is a normal cause of increased contractility?
exercise
how can a ESPVR line be determined?
ESPVR= end systolic pressure-volume relastionship
- determine PV loops of successive decreases in preload
- line created from joining top left corner (point D) of each PV loop over a range of preloads
what is the importance of an ESPVR line?
to describe the maximal pressure that can be developed by a ventricle at any given left ventricle volume
represents end sytolic elastance (mmHg/mL- index of myocardial contrcatility)
insensitive to changes in preload, afterload, & HR
How do the adrenergic agonists work to increase contractility?
norepinephrine & epinephrine stimulate beta1 adrenergic receptors
increase sarcoplasmic Ca2+ concentrations
what pharmacological classes work as positive inotropic agents?
adrenergic agonsit (epinephrine & norepinephrine)
cardiac glycosides (ex. digitalis)
how do the cardiac glycosides increase contractility?
inhibit Na+/K+ pump→ increases intracellular Na+→ no driving force for NCX→ intracellular calcium→ increases contractility
How does sympathetic stimulation increase force of contraction?
- norepinephrine bind beta1 receptors of cardiomyocytes
- activates GCPR
- Galpha-s activates adenylate cyclase
- increases prooduction of cAMP
- activates protein kinase A (PKA)
- phosphorylates targets involved in regulation of Ca2+
- L-type Ca2+ channel→ increased flux
- RyR enhanced Ca2+
- phosphlamban→ release of SERCA2→ faster SR Ca2+ uptake & increased SR content of Ca2+
- troponin I→ relaxes sarcomere quicker→ higher turn over of Ca2+ binding
How does parasympathetic stimulation affect force of contraction?
decreases force of contraction
- ACh binds to muscarinic M2 receptors
- activates GCPR
- Galpha-i inhibits adenylate cyclase
- decreases production of cAMP
- leads to regulatory binding of PKA
- decreased Ca2+ & slower turn around of Ca2+ on sarcomere
- decreased force of contraction
What is the physiological mechanism of action of Ca2+ channel blockers?
- bind to intracellular side of L-type Ca2+ channels
- decrease amount of Ca2+ entering cell
- reduce RyR activity
- reduces amount of Ca2+ released by SR
- decreased contractility of heart
negative inotropic agent
what class(es) of drugs decrease contractility of the heart?
negative inotropic agents: Ca2+ channel blockers & beta blockers
How does sympathetic stimulation influence nodal cells?
- norepinephrine bind beta1 receptors
- activates GCPR
- Galpha-s activates adenylate cyclase
- increases prooduction of cAMP
- activates If (funny current)→ inward Na+ current
- increased depolarization rate of phase 4 pacemaker potential
- IKs increased @ + voltages & deactivates @ more -voltages→ faster (shorter time period) repol.
- phosphorylates L-type Ca2+ channel→ opens at more - voltages & increases Ca2+ influx→ faster upstroke in SA node→ tachycardia
- increase speed of conduction thru AV node
What are the resulting effects of sympathetic stimulation of nodal cells?
- increased frequency of action potentials
- increased propagation of APs thru heart
- shorter duration of ventricular APs
- greater strength of contractility
- faster rate of ventricular relaxation→ shorter duratino of systole (contraction)
How does parasympathetic stimulation influence nodal cells?
ACh
- inhibits adenylate cyclase
- decreasing production of cAMP
- decreasing If activity
- slower depolarization rate of pacemaker potential
- opens up IK-ACh channel
- enhances K+ permeability of SA cells
- hyperpolarizes membrane
- increasing voltage amount to open L-type Ca channels
- generation of fewer action potential/time
BRADYCARDIA
What are the resulting effects of parasympathetic stimulation of nodal cells?
state equation for cardiac output.
CO (L/min)= stroke volume (mL/beat) x HR (beats/min)
what happens to stroke volume if preload is increased?
stroke volume is increased
preload is point B on PV loop
what happens to stroke volume if afterload is increased?
stroke volume decreases
afterload is point D on PV loop
what happens to stroke volume if contractility is increased?
increased slope of ESPVR line
increased stroke volume
state equation for stroke volume
SV= EDV - ESV
EDV= point B on PV loop
true ESV= point D on PV loop
what effect does sympathetic innervation have on heart rate?
increases HR
tachycardia
what effect does parasympathetic innervation have on heart rate?
HR decreases
bradycardia
define stroke volume
volume of blood pumped by one ventricle during one beat
define cardiac output
volume of blood pumped by one ventricle in one minute
what does ejection fraction measure?
fucntion of the left ventricle
the heart’s efficiency or left ventricle health
amount of blood pumped divided by amount of blood that remains in ventricle
state equation for ejection fraction.
EF= SV / EDV= (EDV-ESV) / EDV
normal at rest= 0.55-0.65
what measure is used to determine qualification for heart transplant list?
ejection fraction of 0.15 or less
aka heart is only circulating 15% of blood in ventricle
How can afterload be extracted from PV loop?
pressure at point C
How can blood pressure be estimated from PV loop?
systolic= pressure at peak of phase III
diastolic= pressure at point C
What is stroke work and how can it be obtained?
energy required to eject blood
area w/in PV loop
define barometric pressure.
pressure= weight/force over a surface area
barometric pressure applies the stratosphere, approximately 19km of gas at sea level
what are the convective processes in O2 transportation?
ventilation: atmosphere into lung & circulation to systemic capillaries
when is diffusion utilized in O2 transport?
in the aleolar-capillary membrane & capillaries into cell and mitochondria
these are slow steps of O2 transport
Approximately how much O2 is consumed at rest?
300mL/min or 432L/day
functions of respiratory system
- gas exchange
- talking
- hormonal
- and others
requirements of alveolar-capillary membrane
large & thin
- membrane thickness
- thinner for faster diffusion
- thicker so it won’t break under pressure
- 1-1.5micrometers
- large surface area to maximize diffusion area
- 90m2 (tennis court)
- make it fit by forming vesicular shape
- all need access to atmosphere & to blood flow
- branches 23X
- approximately 200mL in pulmonary capillaries
anatomical dead space
conducting zone
air moves thru by bulk flow, not diffusion
area from trachea down thru terminal bronchioles (16 divisions)
holds approximately 150mL of air constantly
respiratory zone
aka alevolar region
where gas exchange takes place
last 7 divisions
physiological dead space
areas of alveolar region that do not participate in gas exhange
usually due to collapse of alveola or b/c alveolae are not perfused w/blood
technically includes anatomical dead space as well, therefore can only be larger or equal to anatomical dead space
What is the normal adult lung volume after expiration?
3L
5% to dead space volume
dead space volume
5% of lung volume at end of normal expiration
usually 150mL
Where do lung volume changes occur and what is the normal range?
only in alveolar volume
total lung volume= 1.5-7L
tidal volume
VT
volume of breath during normal breathing
approximately 500mL: 150mL to dead space & 350mL to alveolar space AT REST
increases when not quiet breathing
how is an individual’s alveolar surface area calculated?
1m2/kg body mass
average is 70-90m2
what is functional residual capacity?
gas left in the lungs after expiration
approximately 3L
functions of anatomical dead space
- conduct air to gaseous exchange region of lungs
- heat air to body temp (37ºC)
- humidify air to saturation level
- clean air
- cilia in larger bronchioles and above
- muscous glands in brochi
- goblet cells in bronchioles
- defense against microbes
- alveolar macrophages
- IgA & IgM
why does blood pool in the lower extremities when you stand up?
gravity & high venous compliance
How does mean arterial pressure (MAP) change upon standing?
- initial reduction along w/VR & CO due to gravity & high venous compliance
- restored via baroreceptor reflex
- increases HR, contractility, & TPR
- restoring MAP, VR, & CO
how does vascular pressure vary in the supine position?
very little in arterial (head to toe: 95-100-95) or venous (head to toe: 5-2-5) systems
how does blood pressure vary while standing?
0.78mmHg/cm from the heart
bp is reduced above the heart & increased by the same amount below the heart
what are the physical mechanism(s) that oppose blood volume pooling in the venous system?
- one-way valves
- respiratory pumps
- muscle pumps
where is it normal for a negative blood pressure to be found in the body?
the venous system above the heart
how are vascular pressure changes in the standing position calculated?
distance from heart in cm x 0.78mmHg/cm= XmmHg
if in arterial system +/- depending on relative position of artery to heart from the mean arterial pressure (MAP) at the level of the heart
if in venous system +/- depending on relative position of vein to heart from right atrial pressure (RAP)
what is the purpose of the nucleus tractus solitarius?
relay station & integration center for afferent information from multiple organ systems
aides in homeostatis
How does the baroreceptor reflex work?
- decrease in arterial bp
- decrease in afferent firing from baroreceptors (glutaminergic)
- reduced stimulation of NTS (nucleus tractus solitarius)
- reduces inhibition of vasomotor center (GABAergic)
- increases activity in bulbospinal pathway
- decreases activation of caridoinhibitory area
- EFFECT: increase in sympathetic & decrease in parasympathetic innervation
- increase HR, ventricular contractility, CO, & total peripheral resistance→ normalize bp
define orthostatic hypotension w/its causes & effects
inability to restore blood pressure to normal w/change in body position
most likely due to hypovolemic causes of bleeding or dehydration, but may also be due to impaired baroreceptor reflex
may cause syncope
how is orthostatic hypotension tested for?
tilt table test
what happens to blood pressure when standing from a supine position?
- increase in venous volume of 500mL in LEs
- decrease in intrathoracic blood volume (20%)
- decrease SV of 30-40%
- decrease MAP
- activation of baroreceptor reflex or syncope due to decreased cerebral perfusion
How does standing motionless effect BP?
- motionless standing >5min
- increases venous pressure in LEs
- increases venous capillary hydrostatic P in LEs
- increases filtration in LEs
- decreases venous blood volume
- decreases central venous pressure
- decreases VR & CO
- decreases cerebral perfusion
- syncope
how does the skeletal muscle pump reduce increase venous return?
skeletal muscle contraction forces venous blood toward heart
venous valve prevent backflow of blood
reduces blood pooling in LEs, increases VR, reduces venous volume, & venous pressure
how does the thoracic muscle pump reduce venous pooling?
- deep inspiration
- decreases intrathoracic pressure
- decreases central venous pressure & right atrial pressure
- increases driving pressure form LEs to central venous space
- increases venous return= decreases venous pooling
how does the body meet the increased O2 demand seen in exercise?
- increase in blood flow to active muscles
- SNS increases CO
- local vasodilation per metabolic theory increases blood flow to active muscles
- active muscles extract more O2 from blood
- greater blood flow via more capillaries→ greater surface area for exchange
- temperature effect on hemoglobin saturation curves
What are the effects of exercise on the cardiac cycle?
- slow filling phase of diastole significantly reduced
- shortened TP segment on ECG
- 1st/most effected
- cardiac period shortened to 330-500ms
- reduced systolic duration
- atrial contaction maintains EDV & SV
- increases in ventricular & aortic pressures
what happens to stroke volume at very high HRs?
decreases due to reduced ventricular filling time
b/c systolic and diastolic phases are significantly decreased
why does stroke volume increase during initial phases of exercise?
- increased venous return due to increased preload
- increased contractility causes reduced ESV
how does cardiac output increase above 15L/min?
stroke volume plateaus around 160mL/beat
so CO increases due to elevations in HR: which is caused by increased SNS activity & decreased PNS activity
what are normal/trained parameters for HR?
60-80bpm healthy adults
90-100bpm sedentary, middle aged
30s bpm for elite athletes
10W exercise program can reduce HR by 10bpm
what are normal/trained parameters for SV?
- untrained: 60-70mL/beat @ rest & 110-130mL/beat exercising
- elite athletes: 90-130mL/beat @ rest & 150-220mL/beat exercising
- increased filling→ EDV
- greater contractility→ increased ejection fraction→ reduced ESV
what are the major effects of long term aerobic conditioning?
- reduces resting HR (increased vagal tone)
- physiological hypertrophy (SV)
- stimulates angiogenesis to exercising skeletal & heart muscle
How does physical activity affect total peripheral resistance?
- increases sympathetic activity
- increases CO
- increases mean arterial pressure
- induces vasoconstriction
- increases TPR
- increases CO
- locally causes massive vasodilation (active muscles)
- GREATLY reduces TPR
Overall recution in total peripheral resistance
list typical pressure ranges in the heart chambers.
- right atrium: -4 to 4mmHg
- right ventricle: 25/0mmHg
- pulmonary artery: 25/8mmHg
- left atrium: 6-12mmHg
- left vetricle: 120/0mmHg
- aortic artery: 120/80mmHg
what is the physiological origin of S1?
occurs at closing of mitral & tricuspid valves
at the beginning of systole
produced by sudden stretching & subsequent vibration of compliant valves, surrounding connective tissue, & vessels
not by actual valve closure
**timed with upstroke of carotid pulse & beginning of ventricular contraction**
what is the physiological origin of S2?
occurs at closing of pulmonic & aortic semiluniar valves
at the beginning of diastole
produced by sudden stretching & subsequent vibration of compliant valves, surrounding connective tissue, & vessels
not by actual valve closure
what is the physiological origin of S3?
rapid ventricular filling
low pitch sound
most likely heard in apex of the heart or mitral area
diastolic heart sound after S2
normal finding in children, thin adults, thin adults & pregnanct women→ 1st 3 due to thin chest wall
what is the pathological origin of S3?
overfilling of ventricle during rapid ventricular filling phase
ex. aortic or mitral valve regurgitation
what is the physiological origin of S4?
NONE
this is a pathologic sound only
what is the pathological origin of S4?
corresponds to atrial contraction
heard late in diastole jsut before S1
vibrations caused by blood on low compliance ventricular walls
best heard over mitral valve area (apex of heart)
ex. aortic stenosis, ventriular hypertrophy due to chronic HTN
identify auscultation points of heart sounds.
all physicians take money
- aortic valve sounds→ right parasternal 2nd intercostal space
- pulmonic valve sounds→ left parasternal 2nd intercostal space
- tricuspid valve sounds→ left parasternal 4th intercostal space (sometimes 3rd or 5th)
- mitral valve sounds→ left midclavicular 5th intercostal space (apex of heart)
what is the physiological origin of S1 splitting?
left ventricle begins contraction slightly before right
mitral valve closes slightly before right
usually too narrow to hear splitting, but can be completely normal
what is the pathological origin of S1 splitting?
asynchrony of electrical system
ex. bundle branch block
cannot tell from S1 which valve is closing first
what is the physiological origin of S2 splitting?
inspiration
- decreased intrathoracic pressure
- increases venous return & RV filling (RVEDV)
- delays closure of pulmonic valve
what is the pathological origin of S2 splitting?
not heard during inspiration
may occur from conduction abnormalities, congenital defect, or conditions that delay RV emptying
ex. right bundle branch block/ atrial septal defect/ pulmonic stenosis
how are murmurs produced?
turbulent flow of blood
blood flow down large pressure gradient through narrow opening
categorized as systolic or diastolic depending on when they are heard
Causes of isolated systolic heart murmur.
ASS= aortic (or pulmonic) stenosis creates systolic murmur
- aortic stenosis
- larger than normal difference in pressures between aorta & LV (LV higher to open valve)
- sound generated as blood is forced thru narrowed aortic valve
- mitral regurgitation
- blood under high pressure from LV flows thru narrow mitral valve back into LA
- pulmonic valve stenosis
- tricuspid valve regurgitation
when is a systolic murmur heard relative to the other heart sounds?
between S1 & S2
What would cause a S1-ssssss-split S2 heart sound and why?
atrial septal defect
murmur best heard over pulmonic area
- LA pressure exceeds RA pressure→ blood flows L to R
- increased RV EDV
- produces prominent murmur thru pulmonic semilumar valve ejecting excess blood
- delayed pulmonic closure creates split S2
- not during inspiration due to linked atria= no net pressure change
What would cause a S1-ssssssss-split S2 heart sound and why?
ventricular septal defect
murmur best heard over tricuspid area
holosystolic: will be heard from S1 into S2
- LA pressure exceeds RA pressure→ blood flows L to R throughout contraction
- pressure between ventricles is not sufficient to generate audible flow during diastole
- RV sufferes volume overload→ delayed pulmonic closure→ split S2
what can develop if a ventricular defect goes untreated?
irreversible pulmonary HTN
RV hypertrophy
Eisenmenger syndrome: RV hypertrophy exceeds LV muscle mass to reverse ventricular shut→ blood flows right to left during systole
Causes of isolated diastolic murmur.
- aortic regurgitation
- produced by higher pressure blood back flowing thru narrow aortic valve to low pressure ventricle
- mitral stenosis
- larger than normal pressure difference between LA & LV (LA higher to open valve)
- ventricular filling occurs thru narrowed mitral valve
- pulmonic regurgitation
- tricuspid stenosis
when is a diastolic murmur heard relative to the other heart sounds?
between S2 & S1
S1- S2- ssssss-S1
What are the causes & effects of concentric hypertrophy?
- definition: thickened ventricular wall & unchanged or reduced ventricular chamber diameter
- thickening of sarcomere
- causes: increased afterload (pressure overload)
- chronic HTN
- aortic valve stenosis
- effects: decreased ventricular compliance→ S4 heart sound
- heart failure
what are the causes & effects of eccentric hypertrophy?
aka dilated hypertrophy
- definition: increased ventricular chamber diameter w/or w/o heart wall thickening
- sarcomeres lengthen
- reversible
- may produce S3 (phys. or path. origin)
- causes: volume overload (increased preload)
- exercise training
- mitral regurgitation
- aortic regurgitation
- effects: stress on heart→ heart failure
How does aortic valve stenosis affect the PV loop?
- narrowed aortic valve
- high outflow resistance/increase in increased afterload
- point C & D shift up
- impaired left ventricualr emptying- increased ESV
- points A & D (along ESPVR line) shift R
- decreased SV
- phase I is shorter
How does pulmonic valve stenosis affect the PV loop?
increased apparent afterload→ rise in points C & D
decreased SV→ shortened phase I
increased ESV→ points A & D shift right
how does mitral valve stenosis affect the PV loop?
decreased preload (EDV)→ points B & C shift left
decreases stroke volume→ shortened phase I & III
decreases afterload→ lower peak of phase III, points C & D shift down
how does tricuspid valve stenosis affect the PV loop?
decreased preload (EDV)→ points B & C shift left
decreases stroke volume→ shortened phase I & III
decreases afterload→ lower peak of phase III, points C & D shift down
how doe aortic valve regurgitation affect the PV loop?
increases preload (EDV)→ points B & C shift right
increases stroke volume→ lengthened phase I & III
eccentric hypertrophy→ increased compliance
regurgitation backflow→ all points become curves; A/D & B/C volumes are no longer equal
how does pulmonic valve regurgitation affect the PV loop?
increases preload (EDV)→ points B & C shift right
increases stroke volume→ lengthened phase I & III
eccentric hypertrophy→ increased compliance
regurgitation backflow→ all points become curves; A/D & B/C volumes are no longer equal
how does mitral vavle regurgitation affect the PV loop?
increases preload (EDV)→ points B & C shift R
decreased afterload→ lower peak of phase III; points C & D shift down
blood backflow→ decreases ESV→ points A & D shift left
increases total SV, but decreases effective SV
abberant flow causes curvature of all points
what type of murmur does aortic stenosis create?
systolic
between S1 & S2
what type of murmur does pulmonic stenosis create?
systolic
between S1 & S2
what type of murmur does aortic regurgitation create?
diastolic
between S2 & S1
what type of murmur does pulmonic regurgitation create?
diastolic
between S2 and S1
what type of murmur does tricuspid stenosis create?
diastolic
between S2 & S1
what type of murmur does mitral valve stenosis create?
diastolic
between S2 & S1
what type of murmur does tricuspid valve regurgitation create?
systolic
between S1 & S2
what type of murmur does mitral valve regurgitation create?
systolic
between S1 & S2
why is parallel vessel organization advantageous over series organization?
series organization means that all vessels have the same blood flow (L/min)
parallel enables independently regulated blood flow to each organ or region→ adaptability to different conditions
what are the parameters that affect hemodynamics?
- blood vessel diameter: capillaries/vena cava
- mean blood flow velocity: capillaries/aorta
- total cross-sectional area: aorta/capillaries
- blood volume distribution: arterioles/venous branches
- total peripheral resistance/systemic vascular resistance: venous branches/arterioles
- mean blood pressure: vena cava/aorta
define transmural pressure.
pressure measured w/blood pressure cuff
Pi-Po
Pi= pressure stretching the vessel
Po= pressure compressing the vessel
influences vessel diameter
increased Pi→ increased wall tension
increased radius→ increased wall tension
LaPlace’s law
bigger vessel radius→ bigger wall tension required to counter any given transmural pressure
T (wall tension)= Ptm x radius= (Pi-Po)r
which vessel have the greatest wall tension?
aorta
how does LaPlace’s law explain aneurysm progression?
increased radius→ requires increased wall tension→ further thins & weakens wall→ increased radius, etc.
define driving pressure.
pressure difference along length of vessel
responsible for blood flow
deltaP= P1- P2
systemic: P1= aorta & P2= RA
pulmonary: P1= pumonary artery & P2=LA
how is pulmonary driving pressure calculated?
pulmonary vascular resistance= (mean pulmomary artery pressure - pulmonary wedge pressure) / CO
how is systemic driving pressure calculated?
effectively equal to mean arterial pressure (MAP) in systemic b/c RA pressure is so low
MAP= CO x TPR (total peripheral resistance
MAP= diastolic pressure + 1/3 (systolic - diastolic)
how is pulse pressure calculated?
PP= systolic bp - diastolic bp, in mmHg
represents force that the heart generates for each contraction
where is blood flow slowest in laminar flow?
around the edges of the vessel
parabolic shape w/fastest flow in the center
makes relationship between bp & flow proportional
how can the likelihood of turbulence be predicted for a given vessel?
Renolds number, dimensionless index of flow’s tendency to become turbulent
higher #= higher likelihood of turbulence
Re= [diameter(D) x flow rate(v) x fluid density(rho)] / viscosity (eta)
therefore higher diameter, flow rate & density→ turbulence & higher viscosity→ less turbulence
Darcy’s law
flow= (P1-P2)/resistance (R)
- in systemic circulation:
- flow= LV output= CO (L/min)
- P1-P2= aortic pressure-RA pressure= MAP (mmHg)
- R= total peripheral resistance (TPR) or systemic vascular resistance (SVR) (mmHg•min/L)
CO = MAP/TPR
MAP = CO x TPR
TRP = MAP/CO
what determines total peripheral resistance?
Poiseuille’s for laminal flow
resistance = (8 x viscosity x tube length) / (pi x radius4)
greatly reduced by increased radius
what is the difference between parallel and series vessel arrangements in respect to total resistance?
in series, individual/partial resistances are added
in parallel, total resistance is the reciprocal of the sum of the reciprocal of teh individual resistances→ total resistance is always less than single lowest resistance & resistance of arterioles in one area does not greatly alter the total
how can cardiac output be calculated from O2 consumption?
Fick’s Principle
CO = rate of O2 consumptions (mL/min) / ([arterial O2]/mL - [venous O2]/mL)
venous O2 found in RA, RV, or pulmonary artery
what is static compliance & how can it be altered?
physical property of a vessel
determined by connective/elastic tissue presence
altered by age & dz
less elastic tissue→ more compliance
what is dynamic compliance & how can it be altered?
change in vascular tone due to smooth muscle contraction
altered by sympathetic nervous system stimulation of alpha1 adrenergic receptors
Define compliance.
ability of blood vessels to expand & contract passively w/changes in pressure
disten & increase volume w/increasing transmural pressure
compliance = deltaV / delta P
how is compliance shown on a V v. P graph?
the slope of the curve
veins have high compliance at low pressure & low compliance at high pressures
arteries have low compliance at all pressures
veins & arteries have different levels of compliance, so why/how is the saphenous vein used for coronary bypass?
b/c their compliance is similar at high pressures
what is stiffness?
reciprocal of compliance
stiffness = deltaP / deltaV
what happens to the V v. P curve as compliance goes down?
shifts down & right w/loss of steepness to curve
how is blood in the venous reservoir made available?
used during exercise or hemorrhage
- increased sympathetic activity to venous smooth muscle
- venous smooth muscle contracts
- increases venous tone
- decreases venous compliance
- blood reservoir displaced toward heart
- increased VR & CO
where is central venous pressure measured?
in thoracic vena cava near RA
what parameters affect central venous pressure?
- blood volume
- if increased→ increases CVP
- compliance
- if increased→ decreases CVP
how is central venous pressure calculated and why is it important?
delta(CVP) = delta(V) / Cv
Cv= compliance
clinical importance: determinanat of filling pressure→ preload of RV
how does a decrease in cardiac output affect central venous pressure?
- decrease in SV or HR
- decreases CO
- increases venous volume
- increases thoracic blood volume
increases CVP
how does an increase in blood volume affect CVP?
increases venous pressure
increased CVP
how does venous constriction affect CVP?
- decreases venous compliance
- increases VR
- increases thoracic blood volume
incerases CVP
how does standing from a supine position affect CVP?
shift of blood volume to thoracic venous compartment
increases CVP
how does arteriolar dilation affect CVP?
- arteriolar dilation
- increases blood flow from arterial to venous compartments
- increases venous volume
- increases thoracic venous volume
increases CVP
how does muscle contraction affect CVP?
- muscle contraction
- compresses veins
- blood moved toward heart
increases CVP
how does arteriolar constriction affect CVP?
- increase sympathetic innervation
- arteriolar constriction
- reduces venous blood volume
decreases CVP
what maintains blood flow during ventricular diastole?
Windkessel Effect
aortic recoil after the aortic semilunar valve closes
provides additional push to blood→ moves systemically & in coronary arteries
due to static compliance of aorta & large arteries
what parameters can affect pulse pressure?
- SV: increases PP as it increases
- aortic valve stenosis (decreases SV)
- arterial compliance: decreases PP as it increases
- arteriosclerosis
how is wedge pressure measured?
- pulmonary artery balloon catheters (Swan-Ganz) advanced thru RA & RV into pulmonary artery
- measure pulmonary artery pressure
- inflate balloon
- tip of catheter on distal side of occluding balloon measures pressure
how does wedge pressure serve as a substitute for LA pressure?
due to large compliance of pulmonary circulatory system (negligable change)
What cardiac pathologies can directly increase wedge pressure?
mitral valve stenosis or CHF
What is the normal range for wedge pressure?
8-10mmHg
What are the Starling forces and their importance?
determine net movement of fluids across capillary wall
- forces that move fluids out of capillaries
- capillary hydrostatic pressure (PC)
- intersitial colloid osmotic pressure (πi)
- forces that move fluids into capillaries
- interstitial hydrostatic pressure (Pi)
- capillary colloid osmotic pressure (πC): mostly albumin
which Starling forces are usually insignificant in healthy individuals?
interstitial hydrostatic pressure & interstitial colloid pressure
how is net driving force determined & what does it mean?
net driving force = forces out - forces in
= (Pc + πi) - (Pi - πc)
- if + → net fluid movement out of capillary→ filtration
- if - → net fluid movement into capillary→ reabsorption
hydraulic conductivity & reflection coefficient do not change significantly in physiological conditions
why does capillary hydrostatic pressure (Pc) decrease along the length of a capillary?
decrease in resistance
since filtration is greater than reabsorption over capillary length, how does the excess return to circulation?
via lymphatic system
excudate
interstitial fluid containing proteins as consquence of altered capillary permeability
ex. insect bite
transudate
aka ultrafiltrate
interstitial fluid w/o protein
ex. peripheral edema, pulmonary edema, lymphedema
how does decreased arteriolar resistance cause edema?
- decreased arteriolar resistance
- increases capillary hydrostatic pressure
- increases filtration
- edema
how does increased venous resistance contribute to edema?
- increased venous resistance
- increases capillary hydrostatic pressure
- increases filtration
- edema
how does decreased plasma protein concentration contribute to edema?
- decreased plasma protein concentration
- decreases capillary colloid pressure
- decreases reabsorption
- edema
how does decreased lymph drainage contribute to edema?
- decreased lymphatic drainage
- increases interstitial hydrostatic pressure
- edema
what type of edema is seen in right sided heart failure & why?
- right sided heart failure
- backup of fluid in systemic veins & capillaries
- increases systemic capillary hydrostatic pressure
- increases filtration/reduces reabsorption
peripheral edema
what type of edema is seen in left sided heart failure & why?
- left sided heart failure
- backup of fluid in pulmonary veins & capillaries
- increases pulmonary capillary hydrostatic pressure
- increases filtration/reduces reabsorption
pulmonary edema
why type of edema is seen with liver or kidney failure & why?
- liver/kidney failure
- reduced plasma protein production (liver) or loss in urine (kidney)
- decreases capillary oncotic (colloi) pressure
- reduces reabsorption
peripheral edema
what type of edema is seen with lymphatic obstruction & why?
- lymphatic obstruction caused by sx or radiation
- reduced flow from interstitial tissue to lymph
- increases interstitial hydrostatic pressure
- from fluid accumulation in interstitium
- peripheral edema
- increases reabsorption, but exceeds capacity of capillary reabsorption
often unilateral, dependant on cause of lymphatic obstruction
where are the high pressure baroreceptors located?
carotid sinus
aortic arch
renal afferent arteriole
in order of sensitivity
how do baroreceptors detect bp & communicate it to the nucles tractus solitarius?
stretch sensors: increased bp→ more stretch→ increased firing rate
carotid sinus baroreceptor→ glossopharyngeal (CN IX)
aortic arch baroreceptor→ aortic depressor nerve (branch of vagus/CN X)
renal→ long term bp control via renin-angiotensin
where are the low pressure baroreceptors located?
vena cava
right atrium
pulmonary artery
what do low pressure baroreceptors monitor?
venous return
volume receptors
how do the low pressure baroreceptors work?
- increased venous return
- increases stretch of low pressure baroreceptors
- increases afferent signal to nucles tractus solitarius
- regulation of effector organs to reduce volume
when does the carotid sinus nerve fiber?
when the rate of pressure change is maximal
firing frequency is enchanced on upstroke compared to firing frequency on downstroke at the same pressure
what range is the carotid sinus baroreceptor sensitive to?
50/60-200
nerve firing rate is saturated at bp 200
explain how bp is normalized when a rise in bp increases baroreceptor discharge.
- increase in bp
- increases baroreceptor discharge
- via excitatory glutamate on NTS
- nucleus tractus solitarius excites (w/glutamate) caudal ventrolateral nucleus
- stimulates GABAergic inhibitory neurons that act on vasomotor center
- vasomotor neurons produce effect ouput to increase HR, cardiac contractility, & vasoconstriction
- inhibited by GABA
- decreases their normal effectors
NTS also excites (glutamate) neurons in cardioinhibitory center= nucleus ambiguus & dorsal motor nucleus of vagus→ increases vagal innervation→ decrease HR
what is the end result of increased baroreceptor discharge?
- vasodilation
- venodilation→ reduces preload
- decreases bp→ reduces baroreceptor output
- decreases HR→ reduces CO
if high-pressure baroreceptors are so sensitive, how does HTN exist?
baroreceptors are only effective in short-term
long-term/chronic high bp lead to a ‘resetting’ to higher pressure as normal→ do not signal brain that pressure is elevated
what controls bp in long-term?
renin-angiotensin-aldosterone system
the kidneys
How does the renin-angiotensin-aldosterone system work?
- reduced bp at afferent arteriole of kidney
- stimulates release of renin form kidney
- renin cleaves angiotensinogen from liver in circulating blood to angiotensin I
- lung vascular endothelium angiotensin converting enzyme cleaves angiotensin I to angiotensin II
- constricts resistance vessels→ increases systemic vascular resistance, VR→ + MAP
- aldosterone release from adrenal cortex→ increases sodium & fluid retention in kidneys
- stimulates release of antidiuretic hormone from posterior pituitary→ increases fluid retention by kidneys
- stimulates thirst center in brain
how does MAP change during exercise and why isn’t it corrected by the baroreceptor reflex?
MAP increases by 15-30mmHg during exercise
baroreceptor reflex is reversible reset to operate at higher set MAP point to support higher metabolic demands
extent of reset is proportional to intensity of work
What is the valsalva maneuver?
valsalva maneuver is induced when a large breath is taken & attempts to forcibly exhale w/glottis closed for at least 10 seconds→ straining
common during defecation, heavy weightlifting, & childbirth
how can the valsalva maneuver be useful?
as a test for an intact baroreceptor reflex
how does the valsalva maneuver work?
- Phase 1
- high thoracic pressure→ compresses aorta
- increases arterial bp
- decrease HR via baroreceptor activation
- Phase 2
- high thoracic pressure→ reduces VR
- decreases SV & PP
- decreases arterial PP & MAP
- increase sympathetic outflow
- increase HR & peripheral vasoconstriction
- MAP stops decreasing
- Phase 3: stop valsalva maneuver
- decrease to normal thoracic pressure
- decompresses thoracic aorta
- arterial bp falls
- HR increases
- Phase 4
- increase VR→ increase EDV
- increases CO, MAP, & PP
- activates baroreceptor reflex to transient bradycardia
why must heart patients take care not to become constipated?
b/c straining (valsalva maneuver) can kill them
elevated cardiac work & O2 demand→ increased preload in phases 1 & 4 can lead to ischemia & heart attack
3 main causes of shock.
- inadequate cardiac function
- inappropriate vascular tone
- inadequate blood volume
define shock.
severe reduction in blood supply to tissues
cause of hypovolemic shock
shock due to decreased blood volume
secondary to hemorrhage, dehydration, chronic or severe diarrhea or vomiting, or burns→ decreases CVP
cause of septic shock
refers specifically to decrease tissue perfusion resulting in ischemia & organ disfunction
bacterial toxins released during infection
cytokines create large scale inflammatory response→ massive vasodilation, increases capillary permeability, decreases systemic vascular resistance, & hypotension→ reduces tissue perfusion→ tissue hypoxia
**leading cause of death in ICU**
cause of cariogenic shock
reduced CO
secondary to imparied cardiac function
ex. MI
cause of anaphylatic shock
intense allergic reaction→ vasodilator release & subsequent drop in bp→ reduced organ perfusion
define neurogenic shock & its cause
loss of vasomotor tone throughout body
caused by anesthesia, brain damage, or extreme emotional insult
how does the body compensate for hypovolemic shock?
with volume losses of 10-20% only:
- elevation of HR, cardiac contractility, & vascular resistance
- decreased venous compliace
- retorse MAP
- but CO may be low due to reduced SV
What happens when compensatory mechanisms reach their maximum?
decompensated shock
- compensatory mechanisms reach their maximum
- increasing HR & vasoconstriction
- metabolic demands are not met
- cellular ischemia releases vasoactive mediators causing:
- hypotension
- shortness of breath
- acidosis
- cool, pale skin
- mental confusion
What happens when volume losses exceed 20%?
irreversible shock
- if volume loss is >20% or decompensated shock continues w/o tx for 30 minutes
- cannot be corrected
- further deterioration of : circulatory, endocrine, & CNS
- results in organ damage & death
effects of left sided HF
- decreases CO
- increases pulmonary volume→ pulmonary edema
- increases pulmonary capillary hydrostatic pressure
- can lead to right sided HF
- displays left axis deviation
effects of right sided HF
- decreases pulmonary volume
- decreases L heart volumes
- decreases CO
- increases systemic capillary hydrostatic pressure→ peripheral edema & decreases VR
- elevated jugular pulse pressure
- displays right axis deviation
Other than L or R sided HF, how else can cardiogenic shock be categorized?
by when the heart disfunction occurs in cardiac cycle: systole or diastole
define systolic disfunction & possible causes
reduced ability to contract or eject blood
- causes:
- reduced contractility
- increases afterload
- uncontrolled systemic hypertension
define diastolic dysfunction and possible causes
reduced ability to relax or fill ventricle
- causes:
- reduced ventricular compliance (maybe S4)
- reduced chamber volume→ reduces EDV & SV
- reduced preload from stenosed valve
what relates cardiac performance to overall size of an individual?
cardiac index
how is cardiac index calculated?
CI = CO / body surface area (BSA
normal CI= 2.6 - 4.2 L/min per m2
higher in men than women
what is cardiac index useful as?
if it is below 1.8L/min per m2 then pt may be in cardiogenic shock
residual volume
volume at the end of maximal exhalation
approximately 1.5L
expiratory reserve volume
air that is not exhaled during normal breathing
approximately 1.5L to bring total air left in lungs up to 3L
tidal volume
change in lung volume during normal breathing
approximately 0.5L
inspiratory reserve volume
excess volume not taken during normal breathing, but is by maximal inspiratory effort
approximately 2.5L
define capacity in relation to lung volumes.
sum of 2 or more volumes:
residual volume, expiratory reserve volume, tidal volume, inspiratory reserve volume
which lung volumes can be measured by a spirometer?
inspiratory reserve volume
tidal volume
expiratory reserve volume
but not residual volume
how is residual volume measured?
helium diluting method
- helium (or any inert gas) added to spirometer volume
- concentration measured prior to connection to patient
- pt exhales w/maximlal effort
- pt connected to spirometer
- continual breathing until equilibrium of He is reached
- measure new concentration
how is residual volume calculated?
Fi x Vs = Fe(Vs + VL)
- Fi = intitial He fraction
- Vs = spirometer volume
- Fe = He fraction at equilibrium
- VL = residual volume (as long as pt was connected after maximal expiration)
VL = VS [(Fi - Fe) / Fe]
inspiratory capacity
sum of tidal volume & inspiratory reserve volume
functional residual capacity
sum of residual volume & expiratory volume
vital capacity
sum of expiratory reserve volume, tidal volume, & inspiratory reserve volume
largest breath that can be taken
total lung capacity
sum of all lung volumes
TLC = IRV + VT + ERV + RV
what effect does aging have on the lung volumes?
residual volume inceases
total lung volume increases but not as musch as residual
therefore overall vital capacity decreases with age
what are some dz that reduce vital capacity?
- skeletal: kyphoscoliosis
- weak respiratory muscles
- myopathies
- poliomyelitis
- myasthenia gravis
- lung dz
- diffuse pulmonary infiltration
- pulmonary fibrosis
- large pleural effusion
- collapsed lobe/lung
- pulmonary edema
- obstruction
- asthma
- chronic bronchitis
- emphysema
- bronchiestasis
how do obstructive lung dz affect residual volume, total lung volume and their relation to each other?
greatly increase RV
increase TLV
RV/TLV is increased
how do restrictive lung dz affect residual volume, total lung volume and their relation to each other?
decreases RV
decreases TLC
volumes are decreased proportionally→ normal RV/TLC
how is alveolar compliance calculated?
compliance (C) = (delta V) / change in transmural pressure (dPtm)
what are the 2 methods of creating a positive transmural pressure?
increasing inside pressure (Pi)
decreasing outside pressure (Po)
both cause volume expansion
what does the volume v Ptm curve represent?
compliance
As volume increases, compliance….
decreases
how is lung compliance calculated?
CL = (delta V) / (PA - Ppl)
PA = alveolar pressure
Ppl = pleural pressure
how is chest wall compliance calculated?
Ccw = (delta V) / (Ppl - PB)
Ppl = pleural pressure
Pb = body surface pressure
how is total respiratory system compliance calculated?
CRS = (delta V) / (PA - Pb)
PA = alveolar pressure
Pb = body surface pressure
how are lung, chest wall, and respiratory system compliance related to each other?
1/CRS = 1/CL + 1/CCW
when is respiratory system compliance at its lowest?
at total lung capacity & at residual lung volume
when is respiratory system compliance at its highest?
near the functional residual capacity (FRC)
define resting volume of respiratory system
lung volume at which transmural pressure of the respiratory system is zero
lung volume when lung recoil force is equal to chest wall recoil force (b/c they pull in opposite directions)
Is expiration or inspiration an active process?
inspiration
but expiration can be when it goes below FRC
when is lung compliance at its highest?
at volume close to 0
when is lung compliance lowest?
at total lung capacity
where is resting volume of the lung found?
lung volume at which transmural pressure for lung is zero
PA - Ppl = 0
when is chest wall compliance at its lowest?
at residual volume only
there is no decrease in compliance near total lung capacity
where is resting volume of the chest wall found?
lung volume at which transmural pressure for chest wall is zero
Ppl - Pb = 0
approximately 60% of total lung capacity
what type of pathology makes lungs more compliant?
obstructive dz
ex. emphysema, COPD, chronic bronchitis, asthma
volume-pressure curve shifts left & up
expiration is difficult
what type of pathology makes lungs less compliant?
restrictive dz
ex. lung fibrosis
volume-pressure curve shifts to the right & down
inspiration is difficult
when is more muscle work needed for inspiration?
in restrictive dz
ex. lung fibrosis
when is more muscle work needed for exhalation?
obstructive dz
ex. emphysema
factors that influence compliance
- elasticity of tissue
- principle of interdependence of alveoli
- surface tension
- surfactant
what is responsible for the elastic recoil of the lungs?
& geometrical organization of elastin & collagen fibers
explain the principle of interdependence of alveoli.
an alveolus cannot change its volume w/o affect surrounding alveoli
as one increases, compliance of surrounding decreases→ increasing volume of surrounding alveoli
why does surface tension act to decrease lung compliance?
b/c its works to reduce surface area for a given volume of a different medium
lungs are mostly water, which is different than air
occurs at air-liquid interface
behaves functionally like lung recoil force
LaPlace law
P = 2(ST/r)
ST= surface tension
r= radius
following Laplace’s law, why dont’ smaller alveoli collapse?
principle of interdependence of alveoli
surfactant reduces surface tension→ reduces pressure
what are the effects of surfactant?
- decreases surface tension→ increased compliance
- minimizes fluid accumulation in alveoli
- keeps alveolar size relatively uniform during respiratory cycle→ equalizes ventilation amond alveoli
how does surfactant reduce surface tension?
- replaces surface water molecules
- hydrophilic head faces water surface of alveoli
- hydrophobic tail strongly repels water & points toward air
- also prevents surfactant from diving into water
- reduces air-water interface→ greatly reduces surface tension
how does surfactant insufficiency affect lung elasticity & compliance?
increases lung elastic recoil
2-fold decrease in lung compliance
how does surfactant keep alveolar size relatively uniform during the respiratory cycle?
as an avleoli expands, reduction in surfactant density puts a brake on further expansion
thus rerouting gas to another surrounding alveolus
equalizes ventilation among alveoli
how is surfactant cleared?
degraded by macrophages
recycled or destroyed by type II alveolar cells
what is the normal FRC?
functional respiratory capacity
3L
why is pleural pressure negative at FRC?
b/c lung & chest wall are recoiling in opposite directions
how does pleural pressure change throughout the respiratory cycle?
- end expiration (@FRC): -5
- during inspiration: -8
- external intercostal muscles & diaphragm contraction
- end-inspiration: -10
- further decreases bc inspiratory muscles are still contracted
- during expiration: -8
- muscles relax
pressure given in cmH2O
how does alveolar pressure change throughout respiratory cycle?
- end-expiration (@FRC): 0
- during inspiration: -2
- pressure difference from pressure on body surface (0) is driving force for air to enter lungs
- end-inspiration: 0
- during expiration: +2
- lung recoil force creates + pressure in alveoli
pressures in cmH2O
what are the effects of diaphragm contractions during inspiration?
- decreases in Ppl
- negative PA→ creates driving force from atmosphere into lung
- increases Ptm→ further lung distension, covering higher volume
define air flow.
V (dot on top)
air volume passing thru airway in a unit of time
what is the driving force for air flow?
pressure difference between both end of airway
expiration→ PA - Pao; where ao is airway opening
inspiration→ Pao - PA
what device is used to measure air flow?
pneumotachometer
How does the Hagen-Poiseuille law define air way resistance?
Raw = (PA - Pao) / V•
why is the Hagen-Poiseuille equation for resistance not calculable?
b/c alveolar pressure is nearly impossible to measure
How can airway resistance be realistically calculated?
Raw = (8 x eta x L) / (π x r4)
eta=viscosity of medium→ ignored b/c air’s viscosity if negligable
L= length of airway→ ignored b/c relatively constant
r= airway radius
what is the only influencing factor of airway resistance?
radius of airway
small changes cause a huge effect on resistance due to the power of 4
where is total diameter of the airway largest?
beyond the 16th generation in the respiratory zone
this also means that this is the location w/the lowest resistance
why is airway resistance a poor test for a peripheral lung obstruction?
80% of resistance is in the upper airway
branching of lower airway creates overall decrease in airway resistance, but individual bronchioles or alveolar ducts are too small to cause a change in overall resistance
5 factors influencing airway resistance
- density & viscosity of inspired gas
- Partial pressure of CO2
- changes in sympathetic & parasympathetic innervation
- agents: histamine, acetylcholine, thromboxane A2, prostaglandin F2, & leukotrienes LTB4, C4, and D4
- all bronchoconstrictors
- lung volume
how does respiratory air density or viscosity affect airway resistance?
directly proportional
density changes w/altitude
viscosity changed by changing composition of inhaled air: used to replace N2 w/He
so as they decrease so does airway resistance, or vice versa
how does the CO2 partial pressure affect airway resistance?
inversely
decreased airway PCO2→ bronchoconstriction→ increases Raw
important for regional distribution of ventilation: as one region becomes hyperventilated & PCO2 decreases→ airway resistance of that region increases→ diverting ventilation to hypoventilated areas
what effect does sympathetic innervation have on airway resistance?
sympathetic innervation→ bronchodilation via beta-receptors on smooth muscle
as the recoil force increases, compliance…
decreases
why does airway resistance decrease with increasing volume?
lung recoil is distending pressure keeping middle & small airway open
lung recoil increases w/increasing lung volume→ increases airway distending pressure→ airway diameter increases→ resistance decreases
why do emphysema paitents breathe in twice?
once to increase lung volume to decrease the airway resistance
second time to breath for ventilation since reduced resistance makes it easier to breathe
what tests allow physicians to be able to observe changes in airway resistance or respiratory system capacity?
- forced expiratory vital capacity
- spirograms
- flow volume loops
- peak expiratory flow rate
how is forced expiratory vital capacity measured?
- pt takes maximal inspiration
- holds breath
- exhales as hard & fast as possible
- gives:
- forced expiratory volume in 1 sec (FEV1)
- forced vital capacity (FVC)
- ratio of FEV1/FVC→ normal= 80%
- peak expiratory flow rate
define peak expiratory flow rate.
maximum flow rate achieved during forced vital capacity manuever beginning after full inspiration & ending w/maximal expiration
what type of pathologies lead to increases or decreases in FEV1/FVC ratios?
increase→ restrictive dz
decrease→ obstructive
how does obstructive lung dz affect forced vital capacity & forced expiratory volume in 1 second?
FEV1 & FVC are reduced, but unproportionally
FEV1 is reduced much more
so FEV1/FVC is decreased
FVC is decreased slightly b/c some airways are closed during forced expiration
FEV1 decreases b/c of high airway resistance & reduced FVC
how does restrictive lung dz affect forced vital capacity & forced expiratory volume in 1 second?
FEV1 & FVC are greatly reduced due to small total lung capacity
change is proportional or nearly so→ FEV1/FVC is normal or slightly increased
what are the forces acting on or creating alveolar pressure?
lung recoil force
expiratory muscle force
what is spirogram?
plot of exhaled volume as a function of time during a single forced expiration
how are flow volume loops obtained?
- expiratory flow is directly measured during forced expiration & plotted against lung volume
- when different slopes of previous expired volume-time curves are calculated in units of L/sec & plotted against corresponding lung volumes
what parameters are measured by the flow volume loops?
- peak expiratory flow rate (PEFR)
- dependent on pt’s effort
- forced expiratory flow at 75% capacity (FEF 75)
- 25% of vital capacity is exhaled & 75% is in lung
- FEF 50
- FEF 25
why is peak flow, FEF 75, 50, and 25 decreased in restrictive dz?
reduced lung volume
why is peak flow, FEF 75, 50, and 25 reduced in obstructive lung dz?
due to high resistance
what happens to peak flow and FEF 75/50/25 values when compared to total lung capacity in lung dz states?
obstructive: reduced
restrictive: normal
how does obstructive dz shift the flow-volume curve?
shifts to the left w/decreased peak flow rate
b/c increased compliance→ increased residual voume, functional residual capacity, & total lung capacity
how does restrictive dz shift the flow-volume curve?
to the right w/decreased peak flow rate
b/c all lung volumes are proportionally smaller
peak flow rate is reduced b/c vital capacity is smaller than normal
why does restrictive dz cause a problem with inhalation?
increased recoil/decreased compliance→ less driving force for air to enter lungs
why does obstructive dz cause a problem with expiration?
increased compliance/decreased recoil force→ decreased alveloar pressure→ decreased transmural pressure along airway→ equal pressure point w/pleural pressure occurs near alveoli (no cartilage)→ airway collapses→ air is trapped, cannot be expired
how does Pco2 change from atmospheric to physiological circulation?
0 mmHg in atmosphere (low fraction)
40mmHg in alveoli & arteries
43mmHg in mixed venous bllod & metabolic tissue
how does Po2 change from atmospheric to physiological conditions?
150mmHg in atmosphere (20% of 760mmHg)
100mmHg perfused to arteries
0mmHg in metabolic tissue
inverse of Pco2
ventilation
movement of fresh air from ambient to alveoli (site of gas exchange) & subsequent movement of used air back to atmosphere
responsible for refreshing alveolar gas
tidal volume is the sum of ….
dead space volume & alveolar volume
alveolar volume is portion of tidal volume entering alveoli
number of breaths taken in one minute is referred to as?
respiratory frequency FR
what is VE & how is it calculated?
total ventilation
volume of air breathed in a unit of time (usually min)
VE = FR x VT = (FR x VD) + (FR x VA)
respiratory frequncy x tidal volume
how can dead sapce volume be calculated?
where F is gas fractions & P is partial pressures
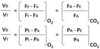
what is basal metabolic rate & its normal values?
minimal value of O2 consumption & CO2 production
250mL O2/min
what are the influencing factors of BMR?
basal metabolic rate
temperature & body size
as they increase, so does BMR
11% for each 1ºC
define Vo2 max
maximum O2 uptake
usually 10x higher than resting value
increases proportional to work load during exercise
what is the respiratory exhange ratio and how does it vary with carbon source?
respiratory exchange ratio (R; lungs) & respiratory quotient (RQ; tissue)
R = CO2 output / O2 uptake
for carbohydrates or glucose (C6H12O6), R=1 due to 1:1 ratio of carbon dioxide to oxygen in: C6H12O6 + O2→ 6CO2 + 6H2O
fats: R=0.7 & proteins: R=0.8
when does R=RQ?
only at rest
when does R not = RQ?
exercise, holding breath, crying, hypoventilation, hyperventilation, etc.
Dalton’s law of partial pressures?
Ptot = P1 + P2 + P3 + …..
ideal gas law
Px x V = Mx x R x T
state the equation to calculate partial pressure of any respiratory gas if given fractional concentration.
Px = Fx (PB - PH2O)
barometric pressure (760mmHg) - water vapor pressure (47mmHg @ 37ºC & 100% humidity as found in lungs)
what factors influence inspired oxygen partial pressure?
inspired oxygen fraction (amount of O2 in supplied air)
barometric pressure
why is O2 partial pressure reduced in alveoli compared to that in the airway or outside?
b/c O2 is continually being removed & entering capillary blood
how is alveolar oxygen partial pressure calculated when O2 is constantly being removed by blood?
PA(O2) = PI(O2) - [PA(CO2) / R]
PA = alveolar partial pressure
PI = inspired partial pressure
what is normal CO2 output?
250mL CO2/min
what is normal alveolar ventilation?
5400mL/min
how is alveolar CO2 partial pressure calculated?

alveolar ventilation equation
VA = [VCO2 / PA(CO2)] x 863
doubling of alveolar ventilation has what effect on arterial CO2 & O2 partial pressures?
CO2: reduction by 1/2
O2: increases
normoventilation
normal alveolar ventilation
Pa(rterial)CO2 =40mmHg
hyperventilation
alveolar ventilation is increased
inexcess of metabolic demand
ParterialCO2 <40mmHg
causes of hyperventilation
- ASA toxicity
- high altitude hypoxia
- progesterone (pregnancy)
- anxiety
- metabolic acidosis
hypoventilation
alveolar ventilation is decreased
not meeting metabolic demand
ParterialCO2 >40mmHg
causes of hypoventilation
- primary cause: barbituate overdose
- secondary cause: compensation for metabolic acidosis
hyperpnea
increased ventilation w/o changes in CO2 partial pressure
ex. exercise
tachypnea
breathing w/increase respiratory frequency
why is increased ventilation during exercise not hyperventilation?
b/c alveolar ventilation increases to keep arterial PCO2 at normal value of 40mmHg
why is there a difference between alveolar (A) and arterial (a) PO2 & what is the normal value?
caused by normal anatomical shunts (pulmonary circulation)
normal difference is 3-10mmHg
Krough’s diffusion constant
K
d x alpha
d= diffusion coefficient
alpha= solubility
define diffusing capacity
DL
d x alpha x (A/T)
- d= diffusion coefficient
- alpha= solubility
- A= area of membrane
- T= thickness of membrane
how is diffusing capacity calculated?
application of Fick’s law for passive diffusion & rearrangement
DL = rate of passive diffusion (Vgas) / (P1 - P2)
why can’t the diffusion capacity for O2 be calculated directly?
b/c pulmonary capillary O2 partial pressure changes from the entrance @ 40mmHg to 100mmHg at teh end of the capillary
what is used to estimate diffusino capacity of O2 and how?
diffusion capacity of CO
DL = VCO / (PACO - PCCO)
but CO is used in very small concentrations, so PCCO=0
only difference between DLCO & DLO2 is due to their respective solubilities (alpha)
how does exercise influence diffusion capacity?
diffusion capacity increases due to recruitment & distension of pulmonary capillaries
better matching of blood flow & ventilation (greater efficiency)
how does body position influence diffusion capacity?
diffusion rate is increased in supine position v. upright
due to increased pulmonary capillary volume & more even distribution of pulmonary blood flow
how does body size influence diffusing capacity?
increases proportionally due to increased surface area that comes w/increased body size
what pathological factors can reduce diffusing capacity?
- increased thickness, ex. pulmonary edema
- decreased surface area
- decreased capillary volume
seen in both obstructive & restrictive pulmonary dz, as well as LV HF & pneumonia
why does the amount of blood entering the pulmonary capillary decrease along its length?
decrease in driving force as the partial pressure reach equilibrium
how fast does gas/blood equilibrium usually take in the pulmonary capillaries?
1/4 to 1/3 of a second
but blood transit time in the capillary is on average 1 second
define diffusion limitation.
incomplete equilibriation of O2 partial pressure between alveolar gas & capillary blood
arterial PO2 remains below alveolar PO2, creating an alveolar-arterial PO2 difference
why is there no diffusion limitation on CO2?
b/c CO2 diffusing capacity is 20x higher than that of O2
when is diffusion limitation seen during exercise?
not at all in healthy subjects at sea level
CO increases much more than pulmonary capillary volume increases→ shortening blood transit time thru lung capillaries
will be seen at high altitudes or exercise of dz individuals
what are the direct causes of diffusion limitation?
low diffusing capacity
caused by decreased alveolar surface area or increased thickess of air-blood barrier
define perfusion limitation.
limitation of gas exhange by the rate of blood flow
O2 diffusion into after equilibrium until 1 second transit time is complete
why is CO the ideal gas for measuring diffusing capacity?
limited almost entirely by diffusing properties
b/c partial pressure in capillary blood is very low due to rapid combination w/hemoglobin
thus biggest influencing factors are air-blood barrier thickness & surface area
pulmonary vascular resistance is ___ times less than total peripheral resistance.
16
as displayed by low driving pressure difference from R ventricle to left atrium
since pulmonary intravascular pressure is low, blood flow is strongly affected by?
hydrostatic (gravity) forces & perivascular pressures
how does perivascular pressure affect large extra pulmonary vessels?
- lie in mediastinum
- mediastinum pressure=pleural pressure= -
- pressure pulls vessels open
- during inspiration, mediastinum pressure is more -
- greater vessel dilation
how does perivascular pressure affect the pulmonary arteries & veins?
surrounded by lung tissue
same as pleural pressure→ pulled open by - pressure→ dilated by further negative pressure during inspiration
how does perivascular pressure affect alveolar capillaries?
lie w/in alveolar wall→ surrounded by alveolar pressure
compressed during inspiration when alveoli are full
how is PVR reduced?
either widening of already perfused vessels or opening of closed vessels
both occur w/increased CO
perfusion is increased 3-6x resting levels w/maximal 2x in pulmonary artery
when is pulmonary vascular resistance lowest?
at functional residual capacity of the lungs
b/c both alveolar & extaalveolar vessels are in their relaxed states
how does increased lung volume affect pulmonary vascular resistance?
increases alveolar resistance as they are compressed
decreases & dilates extraalveolar vessels (greater lung volume means more negative perivascular pressure)
there for PVR is increased
how does decreased lung volume affect pulmonary vascular resistance?
alveolar capillaries decrease in resistance
but extraalveolar vessels increase steeply in resistance
therefore PVR increases
how does the body respond to regional alveolar hypoxia?
- inhibit Kv channels
- depolarizes smooth muscle
- open Ca2+ channels
- alveolar smooth muscle contracts: vasoconstriction
decreased perfusion to hypoxia alveoli & blood diverted to physiologically acitive alveoli
what can augment pulmonary vessel constriction during regional hypoxia?
increased PCO2
hypoxic vasoconstriction in the lungs is due to?
decrease PO2 in the alveolar gas, not the blood
what are the long term effects of global pulmonary hypoxia?
pulmonary hypertension from global pulmonary vasoconstriction all the time
& eventaully right-sided heart failure
what is a possible cause of HAPE?
high altitude pulmonary edema
greater hypoxic vasoconstriction response causing pulmonary hypertension
how does NO affect PVR?
reduces PVR
one similarity to the systemic system
how does teh autonomic nervous system affect PVR?
it doesn’t
how does pulmonary artery perfusion vary throughout the lungs in the upright position?
- top: Palveolar>Ppulmonary artery>Ppulmonary vein→ compressed & no perfusion
- middle: Ppulmonary artery>Palveolar>Ppulmonary vein→ compressed near end of pulmonary capillary & reduced perfusion
- bases: Ppulmonary artery>Ppulmonary vein>Palveolar→ permanently prefused
perfusion increases from peak to base
how does exercise affect lung perfusion in the upright position?
exercise increases pulmonary artery pressure & reduces the inequality of blood perfusion in the lungs
What are the anatomically normal shunts?
- bronchial veins (deoxygenated blood from lung tissue) drains into oxygenated blood in pulmonary veins to the L atrium
- Thebesian veins: coronary venous (deoxygenated) blood draining into L atrium & ventricle
only 1-2% total CO
contribute equally to VA/Q mismatch for normal alveolar-arterial PO2 difference (3-10mmHg)
what are the methods of oxygen transport in blood?
dissolved & chemically bound to hemoglobin
how can the concentration of dissolved oxygen in blood be calculated?
[O2] = solubility of O2 x partial pressure of O2
0.003mL O2/100mL blood for each mmHg partial pressure of O2
thus in normal conditions: 0.003mL O2/100mL blood x 100mmHg PO2 = 3mL O2/1L blood x CO (5L/min) = 15mL O2/min
whereas normal O2 consumption is 300mL O2/min
define O2 capacity
maximum amount of O2 that can be combined w/Hb
what factor(s) is O2 capacity dependent upon?
only Hb concentration
1gm Hb binds 1.34mL O2
normal blood has approximately 15gm Hb/100mL→ 20.1mL O2/100mL
what is the driving force for diffusion of oxygen into cells?
dissolved O2 partial pressure from blood to cells
If diffusion of oxygen to cells depends on O2 partial pressure then how does Hb carried O2 participate?
- dissolved O2 leaves blood for tissue
- lowers partial pressure of O2 in blood
- hemoglobin releases O2 to create equilibrium of partial pressures & further diffusion
why is average O2 saturation of Hb 75% in mixed venous blood?
- amount of O2 delivered to different organs varies
- the body always works w/reserve capacity
what is P50 used for?
P50 =O2 saturation of Hb is 50%
used as a measure for affinity of hemoglobin for O2
what are the advantages of the O2 dissociation curve?
- upper flat portion: even if alveolar oxygen partial pressure falls slightly (dz or altitude)→ loading blood w/O2 only mildly affected
- steep lower portion: peripheral tissues can withdrawl large amount of O2→ only small drop in capillary oxygen partial pressure
assists diffusion of O2 into tissue
how does a decrease in Hb affect O2 saturation & concentration?
reduce O2 concentration
no change in O2 saturation b/c this is dependent on Hb
how does a decrease in oxygen partial pressure affect O2 saturation & concentration?
both would be lowered
what factors increase Hb affinity for O2?
increased pH or decrease in CO2 partial pressure
pH makes a larger difference, but at CO2 concentration affects pH these factors are never isolated in reality
why does low pH reduce Hb affinity for oxygen?
low pH = high [H+]
H+ reversibly binds to imidzole group of amino acid histadine→ conformational change in Hb that decreases affinity for O2
Bohr Effect
how does high CO2 partial pressure affect Hb affinity for O2?
lowers the affinity via:
- CO2 binds N-terminus of alpha & beta chains of Hb
- causes increase in H+ ions→ decreases O2 affinity
how does an increase in CO2 partial pressure aid in oxygen delivery?
- CO2 is made & released to blood stream from tissues
- increased CO2 partial pressure
- decreases Hb affinity for O2
- O2 freely dissolved in blood for diffusion into tissue
where can a double Bohr effect been found?
mother/fetal blood system
how does temperature affect O2 affinity and why is it advantageous or disadventageous?
affinity decreases w/increasing temperature
advantageous b/c temp rises during exercise→ Hb affinity for O2 decreases→ releasing more O2 to tissue when demand is highest
how does 2,3-BPG alter Hb affinity for O2?
2,3-bisphosphoglycerate
reversibly binds beta chains of hemoglobin
increases in 2,3-BPG decrease affinity
helpful in chronic hypoxia (high altitude or lung dz) to unload O2 to peripheral tissue
how does CO interfere w/O2 transport?
- b/c CO & O2 bind the same site on Hb
- CO binds w/240x the affinity of O2
- CO binding to Hb causes an increase O2 affinity→ inhibiting unloading to peripheral tissue
how does tx w/hyperbaric O2 work?
hyperbaric O2 = high O2 partial pressure & would drive CO off O2 binding sites
how is methemoglobin corrected?
methemoglobin has Fe3+ instead of Fe2+
plasma methemoglobin reductase can convert back to normal
Name the forms of CO2 in blood in order of percentages.
bicarbonate
carbamate
dissolved CO2
why is dissolved CO2 considered to play a significant role in its transport?
10% transported in this form
20x more soluble than O2
0.067mL CO2/dL blood per mmHg
where is the majority of bicarbonate formed?
in erythrocytes via carbonic anhydrase
what happens as HCO3- & H+ levels get too high inside the erythrocte?
H+ is buffered by Hb
HCO3- is transported into plasma via exchange protein w/Cl-
what is the Haldane effect?
unloading of O2 in peripheral capillaries facilitates loading of CO2, where oxygenation in the lung has the opposite effect
O2 binding of Hb causes decrease in H+ & CO2 binding of Hb
how are the carbamino compounds formed?
CO2 binds to terminal amino groups in blood proteins
most importantly globin of Hb
how does the CO2 dissociation curve compare to that of oxygen?
- much steeper
- not undergo saturation (doesn’t flatten at top)
- no maximal value for chemically bound CO2
- CO2 concentration will always increase along w/ increase CO2 partial pressure
what factor(s) can decrease CO2 concentrations?
increased temperature
addition or increase in [H+]→ drives H+ + HCO3- ⇒ H2O + CO2 equation to the right
Bohr Effect
increases in CO2 cause an increase in H+ that binds Hb & decreases Hb affinity for O2
what are the types of tissue hypoxia?
- stagnant: low blood flow caused by HF or vascular dz
- anemic: reduced Hb, but Po2 is normal
- histotoxic: inability of cells to uptake or use O2 caused by poisons like CN or HS
- arterial hypoxia: O2 delivery to tissue is decreased by reduction in arterial O2 saturation
what are the types of arterial hypoxia?
aka hypoxemia (ParterialO2 is low)
- low inspired PO2: high altitude
- diffusion limitation
- hypoventilation
- alveolar ventilation/perfusion mismatch
- R to L venous shunt
what types of hypoxia display normal alveolar O2 partial pressure?
- diffusion limited
- V/Q mismatch
- R→L venous shunt
what types of hypoxia have reduced values of both alveolar & arterial oxygen partial pressures?
low inspired oxygen & hypoventilation
what types of hypoxia have an increased alveolar-arterial PO2 difference?
diffusion limited
V/Q mismatch
R to L venous shunt
what can cause diffusion limited hypoxia?
severe lung damage
infections
pulmonary edema
other than arterial hypoxia, what else does hypoventilation cause?
arterial hypercapnia
what drives the correction of hypoventilation?
CO2
why is less ventilation distributed to the apex of the lung in an upright position?
weight of lung stretches alveoli
increases volume & decreases compliance
making it more difficult to achieve change in transmural pressure
how is lung ventilation distributed throughout the lung in the upright position?
lowest at peak & increase toward base
gradient is much less prominent than that of blood perfusion
VA/Q
ratio of alveolar ventilation to blood perfusion
highest at apex & decreases toward base
what is the V/Q status in hyperventilated regions? hypoventilated regions?
V/Q ratio is higher in hyerpventilated & lower in hypoventilated
blood from which area of the lung have the highest PO2?
the upper regions/apex
what is an increased alveolar-arterial PO2 pressure difference a sign of?
decreased efficiency in pulmonary gas exhange
why is arterial-alveolar PCO2 difference caused by V/Q mismatch extremely small & ignorable?
- slope of CO2 binding curve is so steep that increase in CO2 content in arterial blood is not reflected in a significant increase of arterial PCO2
- arterial PCO2 is a strong stimulus for breathing→ small increase in PCO2 enhances ventilation & returns ParterialCO2 nearly to normal
when does V/Q=0?
R to L venous shunt
extreme hypoventilation scenario
alveolar region is not ventilated, but perfused w/blood
what is the V/Q in physiological dead space?
V/Q= infinity
alveolar region is ventilated, but not perfused
steps in differential diagnosis of hypoxia
- measure arterial PCO2
- measure arterial PO2 while breathing 100% O2
- measure diffusion capacity for CO
If arterial PCO2 > normal, it is hypoxia caused by _____.
hypoventilation
If arterial PCO2 < normal, then hypoxia is caused by_____.
low inspired PO2 or high altitude
If arterial PCO2 is normal & arterial PO2 < 400mmHg on 100% O2, then hypoxia is caused by ________.
R to L shunt
If arterial PCO2 is normal & arterial PO2 > 400mmHg, then hypoxia is caused by __________.
diffusion limitation or V/Q mismatch
how are hypoxia caused by diffusion limitation & V/Q mismatch differentiated?
measuring CO diffusion capacity
< normal in diffusion limitation
normal levels in V/Q mismatch
explain how a pt w/bilateral paralysis of the diaphragm is able to breathe
via the use of accessory inspiratory muscles: external intercostals, trapezius, scalenes, sternocleidomastoid
will have tachypnea & show paradoxical inward movement of abdomen w/inspiration
where is the respiratory control center located?
in the medulla & pons
what dictates the effect a spinal cord injury has on breathing?
the spinal origin of the motor neuron to respiratory muscles
where does afferent information to the respiratory center come from?
irritant, mechanical, & chemical receptors
lungs, airway, pharynx, blood vessels, & higher centers
what cell groups comprise the ventral respiratory group?
rostral nucleus retrofacialis, caudal nucleus retroambiguus, & nuclues paraambiguus
why is inspiration a a progressive procress instead of a gasp?
inspiratory signal from the ventral respiratory group is a “ramp” up signal
starts weak & becomes stronger over 2 seconds, then ceases for 3 seconds
what is the main function of the VRG?
basic respiratory rhythm
cardiorespiratory coupling
sympathetic/parasympathetic coupling
bisic activity of bronchial muscle cells
what is the main function of the DRG?
dorsal respiratory group
integration of inputs form airway, lungs, & chemoreceptors
to modify breathing accordingly
where is the DRG located?
w/in the Nucleus Tractus Solitarus
termination of sensory input from vagus & glossopharyngeal nerves w/info from: peripheral chmoreceptors, baroreceptors, & lung respectors (stretch & nocioceptive)
what is the main function of the pneumotaxic center?
controls “off” switch of inspiratory ramp in VRG→ controls respiratory volume
where is the pneumotaxic center located?
in nucleus parabrachialis of upper pons
what are the main pulmonary receptors?
slowly adapting stretch receptors
rapidly adapting stretch receptors
C-Fiber/J-Receptors
muscle spindles
how are the slowly adapting stretch receptors activated?
lung distention (inspiration)
lack of change (holding breath)
deflation of lung below FRC
where are the slowly adapting stretch receptors?
airway smooth muscles
innervated by large myelinated vagal fibers
Explain the Hering-Breuer reflex.
- activation of slowly adapting stretch receptors during inflation
- firing at high frequency
- if inflation maintained, will adapt
- firing at lower frequency
- high activity inhibits further inspiration→ turns “off” inspiration & begins expiration
both inspiratory & expiratory terminating reflex
which receptors resond to irritants?
rapidly adapting stretch receptors
also respond to lung distention
why do lung transplant patients have a diminished cough reflex?
because the cough reflex is controlled by the rapidly adapting stretch receptors
that are located in airway epithelium & innervated by myelinated vagal fibers
vagal fibers connection to the DRG would be severed in a lung transplant
what effect does rapid firing of the rapidly adapting stretch receptors have?
cough reflex
OR
gasp & bronchoconstriction
why does C-fiber activiation cause rapid shallow breathing, bronchoconstriction, & CV depression?
- C-fibers=J-receptors are near capillaries
- innervated by non-myelinated vagal fibers
- activated by increases in interstitial fluid & pulmonary embolism
- increased interstitial fluid causes:
- increases air-blood barrier
- decreases ventilation more so in bases→ shallow breathing is preferable
- bronchoconstriction to divert air to higher ventilation areas
- decreases VR
- CV depression
why is tidal volume & respiratory frequency low in obstructive lung dz?
gamma spindle fibers in muscle tissue respond to amount of motor activty/shortening of inspiratory muscle
in obstructive dz, fibers shorten, but lung volume changes lag behind so fibers shorten beyond normal
this causes large tidal volume which in turn causes low respiratory frquency
where is the central chemoreceptor located?
ventrolateral surface of medulla & medullary raphe
how is the central chemoreceptor stimulated?
by H+ and thus indirectly by CO2
not by hypoxia
how is the central chemoreceptor inhibited?
cold or anesthesia
what dz are cause by defects in the H+ receptors in the central chemoreceptors?
sleep apnea, panic disorder, epilepsy, migraine, sudden infant death syndrome (SIDS)
which peripheral chemoreceptors are not important?
aortic
what is the threshold for carotid body chemoreceptor firing?
PO2 of 80mmHg
although not significant activity until 65mmHg
what cell type is oxygen sensing and where are they located?
glomus or type I cells of the carotid body
explain the theory for how the glomus cells function.
- K+ channel protein is primary oxygen sensor
- inhibition of channel (low PO2 of high [H+])
- depolarization of cell
- activates voltage-gated Ca2+ channels
- Ca2+ enters cell
- causes release of neurotransmitter dopamine
- stimulates nerve endings
what do carotid chemoreceptors respond to?
changes in PO2, PCO2, & [H+]
what creates a more potent stimulus for hypercapnia, acute or chronic exposure?
acute exposure
chronic hypercapnia causes reduced ventilatory repsonse to PCO2
why does acute hypoxia cause decreased ventilation while chronic hypoxia causes increased ventilation?
- acute: decreased O2→ increase firing of peripheral chemoreceptors→ increased ventilation→ lowers alveolar PCO2 & raises alveolar PO2 & causes respiratory alkalosis→ inhibits peripheral & central chemoreceptors→ decrease in ventilation
- chronic: pH is compensated→ no more inhibition of ventilation→ ventilation increases
what is anaerobic threshold?
point in exercise intensity when ventilation is no longer matched w/increase in CO2, causing changes in arterial PCO2, pH, & PO2
muscles produce lactic acid→increases [H+]→ stimulates peripheral & central chemoreceptors→ hyperventilation→ reduces Pco2