11/19- Disorders of Connective Tissue- Skeletal Flashcards
(39 cards)
Describe collagen biochemically
- How many types
- Mutations cause what
- __ bound molecules
- Where are precursor chains made
- Other parts of production
- Collagen bio-synthesis is complex
- More than 16 different types, very abundant in connective tissue
- Mutations in different collagen chains will lead to different diseases where those chains are mostly abundant
- Triple bound molecules, individual precursor chains are synthesized in membrane bound polyribosomes
- Hydroxylated and glycosylated in the rough endoplasmic reticulum (RER)
- Transport, extrusion and proteolysis to remove carboxy (start) and amino (end) terminal propeptide extensions
What are the different types of collagen and where are they found?
- Components
- Diseases associated
Collagen type I is the one we’ll focus on
- a1, a2, and a3
- Found in skin, tendon, bone, and arteries
- Associated with Osteogenesis Imperfecta
Other fun facts:
- Collagen type 2: cartilage and vitreous humor
- Collagen type 3: EDS type IV
- Collagen type 4: found in basal lamina
- Collagen type 5: EDS type I

Type I procollagen is made of what?
- What chromosomes are involved
- 2 pro-alpha 1 chains (chr 17)
- 1 pro-alpha 2 chain (chr 7)
Describe the structure of type I procollagen
- Triple helical structure arranged of tandem Gly-X-Y repeats
- X = proline
- Y = hydroxyproline or hydroxylysine
What is Osteogenesis Imperfecta
- Inheritance pattern
- Genes involved
- Phenotypes
Disorders of collagen and collagen chaperon molecules (post-translation: hydroxylation).
- Most common forms are autosomal dominant.
- Rare recessive: CRTAP, FKBP10, LEPRE-1,PPIB, SERPINF1, SERPINH1, SP7.
- Quantitative defects are associated with milder phenotypes while qualitative defects are more severe.
Describe the basics of OI type I
- Incidence
- Protein involved
- Phenotype
Type I = mild form
- 1/15-20,000
- Type I collagen
- Phenotype
- Multiple recurrent fractures (common an ambulation; steady rate of fractures through childhood and then decrease after puberty; start up again after menopause in women and 60-80 yo in men)
- Normal stature
- Little or no bone deformity
- Blue sclera
- Hearing loss in 50%
- Quantitative defect
What are the type of mutations contributing to OI type I?
Pro-a1 null mutations
- Haploinsufficiency of type I collagen (quantitative defect)
- 1/2 the normal amount of normal type I collagen
What is seen here? What causes it?

Blue/grey sclera in OI type I
- Due to thinning of sclera with color of vessels showing through
What is seen here?

Compression fractures
- Bioconcave appearance of vertebrae (fish-shaped vertebrae)
Describe the severities of type II, III, and IV OI?
- Type II (fatal): lethal in the neonatal period
- Type III (deforming): severe and progressive deformity at birth
- Type IV: mild to moderate bone deformity and variable short stature, common dentinogenesis imperfecta (DI), variable sclerae, and hearing loss
As opposed to type I OI, types II-IV are ______ defects
As opposed to type I OI, types II-IV are qualtitative defects
Describe type II OI
- Incidence
- Gene mutation/consequences
- Prognosis
- Phenotype
- Perinatal, lethal
- Affects 1/20-60,000 (much rarer than type I)
- Mutations in COL1A1
- Glycine substitutions and mutations in the C-terminal pro-peptide (where the protein starts)
- Phenotype: Minimal calvarial mineralization, beaded ribs, compressed femurs, long bones bowing, platyspondyly, small thoracic cage (pulmonary hypoplasia)
- Ultimate cause of death is pulmonary hypoplasia
What is seen here?

OI type II (lethal)
Describe type III OI
- Phenotype
- Qualitative collagen defect.
- Very short stature and bone deformities at birth due to in utero fractures.
- Variable sclerae, dentinogenesis imperfecta (DI), hearing loss.
- Recurrent fractures with minimal trauma + pain.
- Severe deformities with ambulation restriction.
- Adult height 3 ft - 4 ft.
- Pulmonary insufficiency due to severe kyphoscoliosis.
What is seen here?

OI Type III (deforming type)
What is seen here?
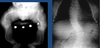
OI type III (deforming type)
Describe the stochiometric effect going on with Pro-a1 in OI types II, III, and IV
- Decreased rate of triple helix formation
- Increased post-translational modification of NH2 terminal to mutation
- Decreased secretion and increased degradation
- Defective collagen fibrils
- Poor mineralization (in bone)
Describe the stochiometric effect going on with Pro-a2 in OI types II, III, and IV
- Biochemical abnormalities similar to the above (a1) but may be less severe
- Phenotype depends on substitution
Why are qualitative defects worse than quantitative?
- Up to 75% of collagen molecules are abnormal
- Abnormal trimers (greater severity than quantitative defects)
- Exporting abnormalities = protein suicide (dominant negative)
Which types of OI are AD?
All of them (I-IV)
What types of mutations are more severe in OI?
“Subtle” point mutations (glycine substitutions) are more severe than large rearrangements which results in null alleles
- Phenotypic gradient from carboxy-terminal to amino-terminal mutations
____ have a ___ effect on normal collagen chains
Mutant collagen chains have a dominant negative effect on normal collagen chains
Mutations in ___affect __% of collagen trimers while _____ mutations affect __% of the trimers
Mutations in COL1A1 affect 75% of collagen trimers while COL1A2 mutations affect 50% of the trimers
What are treatments in OI?
- Supportive
- Interventional
Supportive
- Orthopedic management: intramedullary rods, external braces, splints, castings
- Hearing aids
Interventional
- Infusion of biphosphonates: pamidronate (Aredia®), zoledronic acid (Zometa®)




