15 - Hemoglobin Disorders Flashcards
What are the 2 classes of hemoglobin disorders?
- structural variants
(abnormal globin chain structure due to globin gene mutation; varied clinical effect depending on location and nature of mutation) - thalassemias
(under-production of structurally normal globin chains; generally microcytic/hypochromic anemais of varying severity)
What are the two main globin gene clusters?
- alpha cluster on chromosome 11
(2 alpha, 1 theta, 2 psi, and 2 zeta genes) - beta cluster on chromosome 16
(1 beta, 1 delta, 2 gamma, 1 epsilon gene)

What are the 3 normal hemoglobin species? What are the percentages in a normal adult?
- Hb A (2alpha2beta)
*96% in adults - Hb A2 (2alpha2delta)
*3% in adults - Hb F (2alpha2gamma)
*dominates during fetal life, 1% in adults

What is the incidence of abnormal hemoglobin? What are the usual clinical symptoms?
- 500+ structural hemoglobin variants (mostly single amino acid replacements, occasionally deletions/insertions)
- mostly clinically silent with no functional consequences
What are some examples of clinical consequences caused by abnormal hemoglobin?
- sickling
- Hb instability
- altered oxygen affinity (increased or decreased)
- increased susceptibility to oxidation (to methemoglobin)
- under-production of globin chains
- combinations of the above
What are the 2 main laboratory techniques for diagnosing abnormal hemoglobin?
- electrophoresis (gel or capillary)
- HPLC (high performance liquid chromatography)
Describe hemoglobin electrophoresis
- typically performed in parallel with alkaline and acid buffers
- HbA has isoelectric point of 6.8
- neg charge in alkaline buffers, migrates towards anode (+)
- pos charge in acid buffers, migrates towards cathode (-)

Describe HPLC
- fully automated cation exchange chromatography method
- whole blood method:
- Hb adsorbed onto resin particles
- different species differentially eluted based on affinity for resin by gradually changing ionic strength of elution buffer
- some correlation with migration on alkaline electrophoresis

Describe sickle cell disease
- homozygous abnormality of the beta globin chain
- more common in african americans (1 in 600 homozygous)
- Glu -> Val substitution at AA 6 of the beta chain (beta6val)
- heterozygous HbS “S trait” confers protection against malaria
Describe the pathophysiology of sickle cell disease
- deoxygenated HbS forms long polymers that distort the shape of the cell into an elongated, sickled form
- extend of polymerization is time and concentration dependent
- initially reversible but after multiple sickling/unsickling cycles, membrane damage produces irreversibly rigid sickled cell
- RBC lifespan decreased to 20 days
What affects the concentration of HbS?
- percentage of HbS of total Hb:
- homozygous or heterozygous
- presence of other Hb species (e.g. Hb F)
- total Hb concentration in the red cells (MCHC; Mean corpuscular hemoglobin concentration)
- concentration increased in cellular dehydration
- concentration decreased when co-existent thalassemia
What factors influence the time dependence of sickling?
- transit time of red cells through low oxygen tension microvasculature
- sickling enhanced in anatomic sites with sluggish flow (e.g. spleen and bone marrow)
- blood flow thorugh microvasculature retarded in certain pathologic states (e.g. inflammation)
What clinical settings predispose a patient to sickling?
- hypoxia
- acidosis (shift dissociation curve to the right causing increased deoxygenation of HbS)
- dehydration (hypertonicity causing RBC dehydration)
- cold temperatures (sluggish blood flow)
- infections
What are 2 major effects of RBC sickling?
- chronic hemolysis (correlates with the number of irreversibly sickled cells)
- microvascular occlusion with resultant tissue hypoxia and infarction (related to increased “stickiness” of SS red cells because of membrane damage)
When do symptoms usually begin for a patient with sickle cell?
- newborns clinically fine because of high HbF
- hematologic manifestations begin by 10-12 weeks of age
- clinical severity variable from patient to patient
What are 6 common clinical manifestations of sickle cell disease?
- severe anemia
- acute pain crises (from vaso-occlusion)
- auto-splenectomy (from repeat splenic infarction)
*increases infection risk - acute chest syndrome (major cause of death from pulmonary infections or fat emboli)
- strokes (first usually when 2-8 years old)
- aplastic crisis (acute decrease in RBC production usually from parvovirus B19 infection “fifths disease”)
What does this blood smear show?
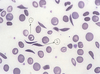
**sickle cell disease
- target cell
- sickled cell
What would the gel electrophoresis results look like for sickle cell disease?
In class she stressed to KNOW THIS!
**bands for S and F hemoglobin (along with the normal A and A2 bands)

What would the HPLC results look like for sickle cell disease?
Large spikes for Hb S and F (note lack of A spike)

What would the HPLC results look like for S-trait?
Large Hb A and S spikes (small F spike, unlike sickle cell disease)

What are the lab findings for HbC disease?
- Hb levels 8-12 g/dl
- numerous target cells
- mild microcytosis
- spherocytes
- occasional C crystals

What would the HPLC results look like for HbC disease?
- >90% Hb C
- NO Hb A!
- <7% Hb F

What are the symptoms of beta-thal major?
- infants well at birth (anemia develops over the first few months of life)
- severe anemia (Hb 2-3 g/dl; recall normal is 12-15)
- virtually all Hb F
- bizarre red cell morphology (hypochormia, targeting)
- transfusion dependent
- severity dependent on adequacy of transfusion program and efficacy of iron chelation

Describe beta-thal minor
- heterozygous form
- mild or no anemia, Hb > 10 g/dl
- microcytosis, scattered target cells
- basophilic stippling
- elevated HbA2 (3.5-7%) **KNOW
- discovered incidentally on CBC (usually asymptomatic)
- incidence:
- common in mediterranean and asian populations
- 1.5% of african americans