Amino Acid Metabolism II Flashcards
(27 cards)
Where do the carbons & nitrogens for cysteine synthesis come from?
What about the sulfur?
- Serine
- Carbon & Nitrogen
- Methionine
- Sulfur
Describe the synthesis and degredation of cysteine
What is the regulatory mechanism in this pathway?
- Synthesis
- Serine reacts with homocysteine (produced from methionine) to form cystathionine via enzyme cystathionine B-synthase & PLP
- Cystathionine is cleaved by cystathionase to form cysteine and alpha-ketobutyrate
- Degradation
- When cysteine is degraded, the nitrogen is converted to urea, the carbons to pyruvate, and the sulfur to sulfate
- Regulation
- when cysteine levels are high, it can inhibit cystathionine B-synthase that is responsible for combining serine & homocysteine

What is cystathioninuria & what are its symptoms?
It is most common in what demographics?
- abnormal accumulation of plasma cystathionine
- Symptoms
- no striking pathological features
- increased urinary excretion
- Demographics
- relatively common in premature infants, as they mature, cystathionase levels rise & levels of cystathionine in the urine decrease (b/c cystathionine is converted to cysteine)
- aduts with a genetic deficiency of cystathionase can causes cystathionuria

What is cystinuria?
Symptoms?
- disorders involving two different transport proteins for cystine (disulfide formed from two molecules cysteine)
- Cystinuria
- defect in the transport protein that permits reabsorption of cystine by renal cells
- cystine, which is not very soluble in the urine, forms kidney stones

Cystinosis is caused by what defective carrier protein?
What does it transport?
Symptoms?
- Carrier protein: cystinosin
- transports cystin across the lysosomal membrane from lysosomal vesicles to the cytosol
- Cystin accumulates int eh lysosomes in many tissues & forms crystals.
- Childen with this disorder develop renal failure by 6-12 years of age, through a mechanism that has not yet been fully elucidated

Where are the defective transport cells for cystinuria & cystinosis respectively?
- Cystinuria
- renal cells
- Cystinosis
- lysosomal cells
Where are the carbons of glutamate derived from?
What happens to glutamate when it is degraded?
Where can glutamate be converted back to glucose?
What amino acids can be converted to glutamate?
- Derivation of Carbons
- Carbons of glutamate are derived form alpha-ketoglutarate via
- transamination
- glutamate dehydrogenase reaction (GDH)
- since alpha-ketoglutarate can be synthesized from glucose, all of the carbons of glutamate can be obtained from glucose
- Carbons of glutamate are derived form alpha-ketoglutarate via
- Degradation
- glutamate is converted back to alpha-ketoglutarate
- transamination
- glutamate dehydrogenase (GDH)
- glutamate is converted back to alpha-ketoglutarate
- Convert to glucose
- in the liver, alpha-ketoglutarate leads to formation of malate
- produces glucose via gluconeogenesis
- in the liver, alpha-ketoglutarate leads to formation of malate
- Amino Acids
- Glutamine, proline, arginine, and histidine contain carbons that can be reconverted to glutamate
- all of these amino acids except histidine can be synthesized from glucose

What enzyme produces glutamine from glutamate?
What is special about this enzyme?
What enzyme converts glutamate back to glutamine?
-
glutamine synthetase
- adds NH4+ to the carboxyl group of the side chain
- one of only 3 human enzymes that can fix free ammonia into an organic molecule
-
glutaminase
- glutamine to glutamate
- ammonia it produces enters the urine

Describe the steps involve in proline synthesis
Describe the steps involved in proline degradation
Which reactions occur in the mitochondria? Which reactions occur in the cytosol?
- Synthesis
- (1) Glutamate is first phosphorylated and then conerted to glutamate semialdehyde
- Semialdehyde spontaneously cyclizes forming an internal Schiff base. This cyclization reaction is nonenzymatic.
- (2) Reduction of this cyclic compound yields proline
- Degradation
- (3) proline is converted back to glutamate semialdehyde, which is (4) oxidized to form glutamate
- Location
- reactions 1, 3, and 4 occur in mitochondria
- reaction 2 occurs in the cytosol

Describe the process of arginine synthesis & degradation
- Synthesis
- Glutamate is is converted to glutamate semialdehyde, which is transaminated for form ornithine
- ornithine is an intemediate of urea cycle & the urea cycle is then used to produce arginine
- Degradation
- arginine is cleaved by arginase to form urea and ornithine

If ornithine is present in excess amounts, it is covnerted to what amino acid?
if ornithine is present in amounts in excess of those required for the urea cycle, it is transmainated to glutamate semialdehyde, which is reduced to glutamate
How is asparate synthesized?
How is asparagine then synthesized from aspartate?
How is asparagine converted back to aspartate?
- Synthesis
- Aspartate is produced by transamination of oxaloacetate
- Asparagine is formed from aspartate by a reaction in which glutamine provides the amide nitrogen of asparagine via the enzyme asparagine synthetase
- Degradation
- Apsaragine is turned back to aspartate via enzyme asparaginase

Why is asparaginase as an anti-tumor agent?
leukemic cells require asparagine for growth
asparaginase acts by converting asparagine to aspartate in teh blood, decreasing the amount of asparagine available for tumor cell growth
How is methionine converted to succinyl Co-A?
- Methionine is converted to S-adenosylmethionine (SAM) which donates its methyl group to form S-adenosylhomocyeteine (SAH)
-
SAH is then converted to homocysteine
- methionine can also be regererated from homocysteine
- Carbons of SAH con be converted to alpha-ketobutyrate, which undergoes decarboxylation to propionyl-CoA
- Propionyl-CoA is then converted to succinyl Co-A

What problems can be caused by high homocysteine levels?
hyperhomocysteinemia
can contribute to arterial damage and blood clots in blood vessels
What are the major functions for the degradation pathways of valine & isoleucine?
- generate energy
- provide precurors to replenish TCA-cycle intermediate (succinyl CoA)
What is the initial step in the degradation of branched chain amio acids?
Second step?
What is the final product of the degradation of these branched chain amino acids? Are they glucogenic or ketogenic?
Where in the body is activity of these reactions the highest?
- Initial Step: transamination
- Valine : alpha-ketoisovalerate
- Isoleucine : alpha-keto-B-methylvalerate
- Leucine : alpha-ketoisocaproate
- Second step: oxidative decarboxylation by alpha-keto acid dehydrogenase
- alpha-ketoisovalerate : isobutyryl CoA
- alpha-keto-B-methylvalerate : 2-methylbutyryl CoA
- alpha-ketoisocaproate : isovaleryl CoA
- Final product
-
Valine (glucogenic)
- Propionyl CoA –> Succinyl CoA
-
Isoleucine (glucogenic & ketogenic)
- Propionyl CoA –> Succinyl CoA
- Acetyl CoA
-
Leucine (strictly ketogenic)
- Acetyle CoA
- Acetoacetate
-
Valine (glucogenic)
- Highest level of activity in the muscle tissue
What are the symptoms of maple syrup urine disease (MSUD)?
What is the biochemical cause of MSUD?
- Symptoms
- poor feeding
- vomiting
- lethargy
- delayed development
- In MSUD, the branched-chain alpha-keto acid dehydrogenase that decarboxylates the branched chain amino acids is defective. As a result, the branched-chain amino acids and their alpha-keto analogs accumulate. They appear in the urine, giving it the odor of maple syrup.
- accumulation alpha-keto analogs is toxic to the brain and other organs

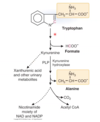
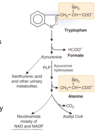
What are teh 7 keogenic amino acids? This means they produce what?
Which are also glucogenic?
- Ketogenic: produce acetyl-CoA or acetoacetate
- Phenylalanine
- Tyrosine
- Leucine (strictly ketogenic)
- Lysine (strictly ketogenic)
- Isoleucine
- Threonine
- Tryptophan
- Also glucogenic
- isoleucine
- threonine
- phenylalanine
- tyrosine
- tryptophan

Describe the degradation of pheynylalanine and tyrosine
- Phenylalanine is hydroxylated to form tyrosine by phenylalanine hydroxylase
- Tyrosine is converted to p-hydroxyphenylpyrivate by tyrosine aminotransferase
- p-hydroxyphenylpyrivate is convrted to homogentisate
- Through two reactions, one by homogentisate oxidase and one by fumarylacetoacetate hydrolase, homogentisate is converted to either fumarate or acetoacetate

PKU is the result of a deficiency in what enzyme?
Symptoms?
How do you treat PKU?
What is the concern with a person with PKU giving birth?
How do we screen for PKU?
- PKU
- phenylalanine hydroxylase
- Symptoms
- build up of pheynlalanine to harmful levels can cause intellectual disability
- musty or mouse-like odor
- lighter skin and hair than unaffected family members
- skin disorders such as eczma
- Treatment
- low protein diet / phenylalanine restricted diet
- babies born to mothers who have PKU have a significant risk of intellectual disability becasue teya re exposed ot very high levels of phenylalanine before birth
- Screen
- spores of B. subtilis are place on agar plate containing B2-thienylalanine (inhibitor of B. subtilis growth)
- Blood sample from infant at lease 24 hrs old is place on agar plate.
- If phenylalanine content of the blood is >2 to 4 mg/dL, the phenylalanine will counteract the effects of the B2-thienylalanine, and bacterial growth will occur
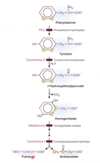
Tyrosinemia -II is caused by deficiencies in what enzyme?
Symptoms?
Treatment?
- Deficiency
- tyrosine aminotransferase
- Symptoms
- lesions of the eye & skin
- neurologic problems
- Treatment
- diet low in tyrosine and phenylalanine
Alkaptonuria is caused by deficiency in what enzyme?
Symptoms?
- Deficiency
- homogentisate oxidase
- Symptoms
- dark urin on exposure of air (b/c autoxidation of excreted homogentisate)
- arthritis and connective tissue pigmentation

Tyrosinemia-I is caused by a deficiency in what enzyme?
Symptoms?
Treatment?
- Deficiency
- fumarylacetoacetate hydrolase
- Symptoms
- liver failure
- cabbagelike body odor
- death within first year of life
- Treament
- diet low in tyrosine & phenylalanine