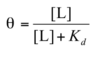Ch 4 - Ch 6 Flashcards
4) The three-dimensional structure of proteins 5) protein function 6) enzymes (116 cards)
Peptide Bond
a peptide bond is a resonance hybrid of two canonical structures
- the peptide C–N bond is somewhat shorter than the C–N bond in a simple amine due to their partial double-bond character
the resonance causes the peptide bodes
- to be less reactive compared to esters
- to be quite rigid and nearly planar
- to exhibit a large dipole moment in the favored trans configuration
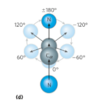
dihedral angles
the polypeptide is made up of a series of planes linked at alpha carbons
the dihedral angle is the angle at the intersection of two planes
peptide conformation is defined by three dihedral angles: phi, psi, and omega
some phi and psi combinations are very unfavorable because of steric crowding of backbone atoms with other atoms in the backbone or side chains
some phi and psi combinations are more favorable because of the chance to form favorable H-bonding interactions along the backbone
Ramachandran plot
shows the distribution of phi and psi dihedral angles that are found in a protein
shows the common secondary structure elements
reveals regions with the unusual backbone structure
dark blue areas represent conformations that involve no steric overlap and the white regions are conformations that are not allowed due to steric constraints

What are secondary structures?
secondary structure refers to any chosen segment of a polypeptide chain and describes the local spatial arrangement of the polypeptide backbone
common, regular arrangements: alpha-helix, beta-sheet, beta-turn
irregular arrangement: random coil
a-helix
- stabilized by hydrogen bonds between amides of an n and n+4 amino acids; h-bonds between the H on the nitrogen atom of a peptide linkage and the carbonyl oxygen atom of the fourth amino acid on the amino-terminal
- 3.6 residues per turn = 5.4 Å
- peptide bonds are aligned roughly parallel with the helical axis
- side chains point out and are roughly perpendicular with the helical axis
- right-handed alpha-helix is the common form
- large macroscopic dipole moment; negatively charged residues often occur near the positive end of the helix dipole

b-sheet
- sheet-like arrangement of backbone stabilized by hydrogen bonds between adjacent segments that may not be nearby
- side chains protrude from the sheet alternating in up and down direction
- parallel or antiparallel orientations are possible
Which amino acid residues are helix formers and which are helix breakers?
Ala and Leu are strong helix formers
Pro and Gly are helix breakers
with Pro, rotation around the N-Calpha bond is impossible
with Gly, the tiny R-group supports other conformations
attractive or repulsive interactions between side chains 3-4 amino acids apart will affect the formation (charge, bulk, and shape)
parallel b-sheets
the H-bonded strands run in the same direction (weaker H-bonds)
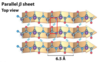
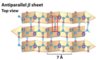
antiparallel b-sheets
the H-bonded strands run in opposite directions (stronger H-bonds)
ß turns
connect the ends of two adjacent segments of an antiparallel ß sheet
the 180º turn involves 4 amino acids; the turn is stabilized by a hydrogen bond from carbonyl oxygen to amide proton in the 4th residue
proline in position 2 (more common) or glycine in position 3 are common in ß turns
Pro: can be in the cis conformation
Gly: because R-group is small, so flexible
often found near the surface of a protein where the two central amino acid residues in the turn hydrogen-bond with water
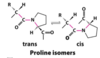
proline isomers
most peptide bonds not involving proline are in the trans configuration (> 99.95%)
about 6% of proline peptide bonds are in the cis configuration. most of the 6% involve ß turns
proline isomerization is catalyzed by proline isomerases
What is the length of a polypeptide with 80 amino acid residues in a single, continuous alpha-helix?
120 Å
an idealized alpha-helix has 3.6 residues per turn and the rise along the helical axis is 5.4 Å
Circular dichroism spectroscopy (CD)
a measurement of the difference in absorption of left-handed and right-handed circularly polarized light
The light-absorbing entity (chromophore) is the peptide bond; a signal is obtained when the peptide bond is in a folded environment
The alpha-helix and b-conformations have characteristic CD spectra (see figure)
a method for assessing common secondary structure
- can see if proteins are properly folded
- estimate the fraction of the protein that is folded in either of the common secondary structures
- monitor transitions between the folded and unfolded states

Tertiary Structure
- stabilized by numerous weak interactions between amino acid side chains
- largely hydrophobic and polar interactions
- can be stabilized by disulfide bonds
- two major classes: fibrous and globular (water- or lipid-soluble)
What are the different types of fibrous proteins?
a-Keratin (a-helix)
Silk (beta conformation)
collagen (triple helix)
all fibrous proteins have high concentration of hydrophobic amino acid residues both in the interior and on the surface of the protein
Keratin
right-handed a-helix crosslinked by disulfide bonds
two a-keratin strands oriented in parallel are wrapped around each other to form a left-handed coiled-coil
tough, insoluble protective structures of varying hardness and flexibility
found only in mammals (hair, wool, nails, claws, horns, hooves, and the outer layer of skin)
chemistry of perms: reduce cystine residues to break disulfide bonds, hair is manipulated (straight or curled), then oxidized to reform disulfide bridges
silk
fibroin is the main protein in silk from moths and spiders
antiparallel beta-sheet: small side chains (ala and gly) allow close packing of sheets that are stabilized by hydrogen bonding and van der Waal’s interactions
extremely strong (stronger than steel), soft, flexible filaments
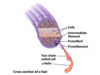
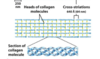
collagen
three pro- and gly-rich left-handed collagen helices intertwine into a right-handed superhelical triple helix
many triple helices assemble into a collagen fibril
in tendons, cartilage, cornea, and bone matrix
high tensile strength (higher than a steel wire of equal cross-section), without stretch
Scurvy and Vit C
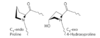
scurvy is caused by a lack of Vitamin C (ascorbate)
Vitamin C is required for the hydroxylation of proline and lysine in collagen; hydroxylation forces the proline ring into a conformation that allows more hydrogen bonds between the three strands of collagen
without ascorbate, degeneration of connective tissue (hemorrhage, tooth loss, poor wound healing and reopening of old wounds, bone pain and degeneration, and eventually heart failure)
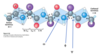
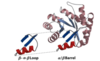
What kind of tertiary structures are in globular proteins?
- proteins are made of different motifs folded together
- motifs (folds): the specific arrangement of several secondary structures
- beta-a-beta loop
- alpha/beta-barrel
- can be found as reoccurring structures in numerous proteins
- domains are an assembly of motifs that can stand alone, often with a separable biological function
x-ray crystallography
Steps needed
- purify the protein
- crystallize the protein
- collect diffraction data
- calculate electron density; regions of greatest electron density reveal the location of atomic nuclei
- fit residues into density
Pros: no size limits; well-established
Cons: difficult for membrane proteins; cannot see hydrogens
The first globular protein structure to be determined by x-ray diffraction was that of myoglobin.
What are the three protein structure methods you need to know?
1) circular dichroism spectroscopy (CD)
2) x-ray diffraction
3) NMR
NMR
steps needed
- purify the protein
- dissolve the protein
- collect NMR data
- assign NMR signals
- calculate the structure: computer generates a family of closely related structures that represent the range of conformations consistent with the distance constraints
Pros: no need to crystallize the protein; can see many hydrogens
Cons: difficult for insoluble proteins; works best with small proteins
intrinsically disordered proteins
- contain segments that lack definable structure; lack hydrophobic core
- composed of high concentrations of Lys, Arg, Glu, and Pro, which force less-defined structure
- disordered regions can conform to many different proteins, facilitating interaction with numerous different partner proteins
- PONDER (predictor of natural disordered regions): the higher the PONDR score (0 - 1.0) the more intrinsic disorder