Exam 2: Biochemistry Flashcards
(286 cards)
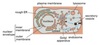
Cell
Compartmentalization
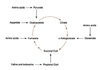
Topologically equivalent spaces:
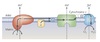
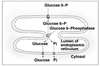
Nucleus & Cytosol
Perinuclear cistern, ER cisterna, Golgi cisterna, Lysosomes, Transport vesicles & Endosomes
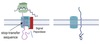
Movement between topologically inequivalent spaces requires translocators.
Cellular Transport
Mechanisms
- Gated transport
- Transmembrane transport
- Vesicular transport
Gated Transport
Large openings act as selective gates.
Between topographically equivalent spaces ⇒ does not cross a membrane.
Ex. nucleus ↔︎ cytosol
Transmembrane Transport
- Between topologically inequivalent spaces ⇒ crosses a membrane
- Uses translocators
- dependent on targeting signals
- protein moved in a denatured form
- Ex.
- import of nascent peptides into RER
- import from cytosol into mitochondria and peroxisomes
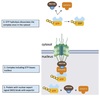
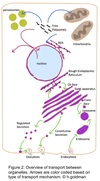
Vesicular Transport
- Between topologically equivalent spaces when each is membrane bound
- Transport vesicles carry proteins and membranes
-
Anterograde → “forward”
- ER to Golgi
-
Retrograde → “backward”
- Golgi → ER
- Endosomes → Golgi
- Ex.
- ER → Golgi
- Golgi → lysosomes / plasma membrane
- Endocytosis & Exocytosis

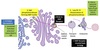
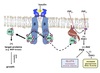
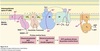
Vesicle Structure
- Inner layer formed from adaptor proteins links outer layer (cage) to the membrane
-
Cage proteins cover cytosolic surface forming a coat
- Assembly requires energy & GTP binding proteins
- Functions:
- collect specific membrane and soluble cargo
- direction formation of vesicles
- Removed before vesicles fuse
- Differs depending on destination and direction of movement
COPII Coating
Coats anterograde transport vesicles from ER → Golgi.

COPI Coating
Coats retrograde transport vesicles from the Golgi → ER.

Clathrin Coating
Transport vesicles from Golgi → endosomal compartments / plasma membrane.
Transport vesicles from plasma membrane → endosomes.

Vesicle Movement
Budding and targeting uses movement along cytoskeletal tracks:
Microtubules using kinesin and dynein
Actin filaments using Myosin II or Myosin V
Motor proteins recruited by Rab proteins.
Vesicle Targeting
Docking and fusion mediated by SNARE proteins.
SNARE proteins on transport vesicle bind complementary SNARE proteins on the target membrane.
Forces two membranes close together so lipid bilayers can fuse.

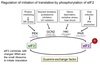
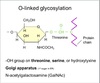
Genetic Code
Definition
The sequence relationship between the bases in the gene or mRNA and the amino acid in the protein.
3 consecutive nucleotides on mRNA ⇒ codon
Genetic Code
Characteristics
- 64 possible combinations of the 4 bases
- 61 code for AA
- 3 code for stop codons
- Codons are contiguous and do not overlap
- Degenerate ⇒ multiple codons can code for a single AA
- Unambiguous ⇒ each codon codes for one AA
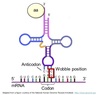
Wobble Hypothesis
The pairing between the codon and anticodon adheres to the usual base-pairing rules at the first two bases but is less strict for the third.
- First two bases predominate in tRNA selection
- Some tRNAs can bind to more than one codon that codes for the same AA
Start Codon
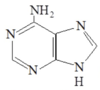
AUG ⇒ Methionine
N-terminal methionine formylated in prokaryotes ⇒ fMet
Stop Codons
UAA ⇒ U are awful
UAG ⇒ U are gross
UGA ⇒ U go away
Rules of
Protein Production
- Anticodon of tRNA pairs with codon of mRNA in anti-parallel fashion
- mRNA read in 5’ ⇒ 3’ direction
- Codons read sequentially by charged tRNAs
- Proceeds from N-terminus ⇒ C-terminus
Protein Synthesis
Stages
- Activation of amino acid
- Chain initiation
- Chain elongation
- Chain termination
- Co/post translational processing
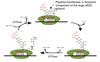
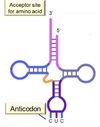
tRNA Structure
Acceptor end links the 5’ and 3’ ends forming clover structure.
3’-OH terminal CCA has AA attached to acceptor site.
Anticodon triplet base pairs with mRNA codon.
Amino Acid
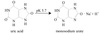
Activation
“Charging the tRNA”
Catalyzed by aminoacyl-tRNA synthetase.
- 20-different aminoacyl tRNA synthetases recognizes ONE amino acid and ALL its cognate tRNAs
- Traps energy from hydrolysis of ATP → AMP + PPi in AA~AMP complex
- Two high energy bonds from ATP required
- Forms high energy bond between tRNA and AA used later to link AA to polypeptide chain
- Proofing mechanism to ensure correct AA attached
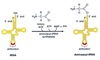
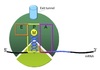
Ribosome Structure
Each subunit contains 3 critical sites:
A site ⇒ accepts the incoming aminoacylated tRNA
P site ⇒ holds tRNA and has ribosomal peptidyl transferase to form peptide bond
E site ⇒ temporarily holds deacylated tRNA until it exits the ribosome
Small Ribosomal Subunit
Functions
- Formation of the initiation complex
- Decodes the genetic information i.e. reads mRNA
- Binds both the 5’ end of mRNA and the tRNA-amino acid complex at the loading site
- Controls the fidelity of codon-anticodon pairing
Large Ribosomal Subunit
Functions
- Contains ribosomal peptidyl transferase activity that joins the AA to the polypeptide chain
- Contains translocation domain
- Contains tunnel where nascent peptide threaded
Translation Initiation