Glycoaminoglycans Flashcards
(25 cards)
What are GAG? proteoglycans? glycoproteins?
- GAGs: Negatively charged heteropolysaccharides
- Proteoglycans: Carbohydrates (GAGs; >95%) + Proteins
- Glycoproteins: Carbohydrates(small amount) + Proteins
- Long, unbranched polysaccharides containing a repeating disaccharide unit. [acidic sugar-amino sugar]n
- Disaccharide units contain
-
Amino sugars
- N-acetylglucosamine (GlcNAc) or
- N-acetylgalactosamine (GalNAc)
** - Acidic sugar **
- Uronic acids
- Glucuronic acid
- Iduronic acid
What are the features and functions of gag?
- Negatively charged and bind to large amount of water (hydrated); extended in solution
- Produce gel-like matrix- forms the basis of ground substance which along with fibrous structural proteins forms the extracellular matrix (ECM)
- Hydrated GAGs provide flexible support to the ECM
- Acts as a molecular sieve in ECM
- “Slippery” consistency of mucous secretions & synovial fluid due to the due to the large number of negative charges on GAGs; they repel each other & slide past each other
- When GAGs are compressed, water is ‘squeezed out’; when compression is released, GAGs spring back to their original hydrated volume
- Contributes to the resilience of synovial fluid and the vitreous humor of the eye
What are the types of GAG ( mucopolysaccrides) and where are they located?
- Located primarily
- On the surface of cells
- In the extra cellular matrix (ECM)
- Mucus secretions - Types:
- Hyaluronic acid
- Dermatan sulfate
- Chondroitin sulfate
- Keratan sulfate
- Heparan sulfate
- Heparin
HYANDURONIC ACID
Composition: D-glucuronic acid + N-acetyl glucosamine
(GlcNAC)
- Non sulfated & Not covalently linked to proteins
- Location: Synovial fluid, vitreous humor, umbilical cord
ECM of loose connective tissue
Functions: Lubricant and shock absorber, role in cell migration during embryogenesis
Dermatan sulfate
Composition: L-iduronic acid + N-acetyl galactosamine 4-S
(GalNAc)
- Location: Skin, blood vessels, heart valves
- Functions: Constituent of skin, role in wound healing
Chondroitin 4- and 6-sulfates
Composition: D-glucuronic acid + GalNAc-4- or 6- sulfate
- Form proteoglycan aggregates with Hyaluronic acid
- Most abundant GAG in the body
- Location: Cartilage, tendons, ligaments, aorta, cornea
- Function: In cartilage it binds to collagen and hold fibers in a tight strong network
- Composition: D-glucuronate-2-sulfate (10%)
(or iduronate-2-sulfate (90%) \+ N-sulfo-D-glucosamine-6-sulfate - (heparans have less sulfate than heparins)
Ø Heparin: - Only intracellular GAG àmast cells lining arteries in liver, lungs and skin
- Anticoagulant: Heparin activates antithrombin III, which in turn inhibits thrombin & other clotting factors
Heparan sulfate
Ø: Basement membrane and cell surfaces
It binds specifically to lipoprotein lipase present in capillary walls
Karatin sulfate
- Composition: Galactose + GlcNAc-6-sulfate
No Uronic acid
- Most heterogeneous as may also contain L-Fucose,
N-acetyl neuraminic acid (NANA) & mannose
- Location: Cornea, bone, cartilage aggregated with Chondroitin sulfates
- Function: In cornea both Keratan sulfate & dermatan sulfate lie between collagen fibrils & facilitate corneal transparency.
Explain the structure of proteoglycans
The protein cores of proteoglycans are rich in Serine & threonine residues, which allows multiple GAG attachments
üMany such Proteoglycans monomers aggregate on a molecule of Hyaluronic acid to form proteoglycans aggregates through ionic interactions and stabilized by linker proteins
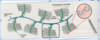
Explain how the the GAG is attached to the protein core
GAGs extend perpendicularly from the core in a bottle brush-like structure
Linkage of GAGs to protein core involves a specific trisaccharide, two galactose residues and a xylulose residue
(GAG—GalGalXyl-O-CH2-protein).
The trisaccharide linker is coupled to the protein core through an O-glycosidic bond to a Serine residue in the protein

Explain the synthesis of glucuronic acid for GAG formation

Where isthe synthesis of GAG, what are the amino and acidic sugars involved? Where does the synthesis of the core protein take place?
- Synthesis of GAG: Golgi, glycosyltransferase
- Amino sugars (amino group donated by glutamine)
- N-acetyl glucosamine & N-acetyl galactosamine
- Acidic sugars
- D-Glucuronic acid & L-Iduronic acid
- Glucuronic acid synthesized by uronic acid pathway
- (3’phosphoadenosyl-5’-phosphosulfate) PAPS is the donor of sulfate group
- Amino sugars (amino group donated by glutamine)
- Synthesis of core protein:
- RER
What is the disease associated with the synthesis of GAG and proteoglycans?
Chondrodystrophies:
- Autosomal Recessive
- Defect in the sulfation of GAG chain
- Improper development and maintenance of the
skeletal system
How are GAG’s degraded?
Degradation of GAG: Lysosomes, Acid Hydrolases
Extra cellular GAG
¯
phagocytosed
¯
fused with a lysosome
¯
endoglycosidases
¯
desulfated & deacetylated
¯
further action of acid hydrolases
What are mucopolysaccharidoses?
- Hereditary disorders caused by the accumulation of GAG in various tissues due to the defect in the lysosomal hydrolases of GAG catabolism
- Progressive disorders, cause skeletal & ECM deformities, intellectual disability
- All of these disorders, except Hunter’s syndrome, are inherited in an autosomal recessive manner
- Homozygous children apparently normal at birth, gradually deteriorate, may die in childhood
- Incomplete GAG degradation results in excretion of oligosaccharides in urine, can be used for diagnosis
- Diagnosis confirmed by assay of lysosomal hydrolase
- Bone marrow, cord blood transplants, enzyme replacement therapy available
Hurler’s disease
Autosomal recessive
Deficiency of a-L-Iduronidase
Degradation of dermatan & heparan sulfate are affected & they accumulate
Mental retardation & corneal clouding
Dwarfing, coarse (dysmorphic) facial features)
Upper airway obstruction, hearing loss
Deposition in coronary artery leading to ischemia and early death
Therapy: Bone marrow or cord blood transplantation and enzyme replacement
Hunters disease, sanflippipo syndrome, Maroteaux-Lamy syndrome ,Morquio’s syndrome
Sly syndrome
Hunter’s syndrome
X-linked recessive
Deficiency of Iduronate sulfatase
degradation of dermatan & heparan sulfate affected
Clinical features similar to Hurler syndrome, but no corneal clouding
Therapy: Enzyme replacement
Sanfilippo syndrome (Types A to D)
Severe nervous system disorders due to defective degradation of heparan sulfate, developmental disability
Maroteaux-Lamy syndrome
Defective degradation of dermatan sulfate
Morquio’s syndrome
Defective degradation of keratin & chondroitin sulfate
Sly syndrome b-glucuronidase deficiency
What are glycoprteins and what are their function?
•Proteins with covalently attached oligosaccharides
•Predominant sugars à glucose, galactose, mannose, Fucose, GalNAc, GlcNAc and NANA
•Functions
Structural molecule: Collagen
Lubricant & protective agent: Mucins
Transport: Transferrin, Ceruloplasmin
Protective: Immunoglobulins
Cell surface recognition by other cells, hormones, viruses
Hormones: hCG, TSH
Antigenicity (blood group antigens)
Enzyme: Alkaline phosphatase
Plasma proteins (except albumin)
What are the linkages between the sugars and peptides?
What is the differnce between proteoglycans and glycoproteins?
**proteoglycans: **
Contain long polysaccharide chains
Repeating disaccharide units in GAGs
Unbranched sugar chains
High carbohydrate content
Glycoproteins
short polysaccride chains
no repeating units, branched sugar units, and low carbohydrate content
How are glycoproteins synthesized in the RER and in the golgi?
- protien sythesis begins and the polypeptide chain is extruded into the RER. 2. A branched oligosaccride is synthesized on dolichol pyrophosphate 3. the oligosaccride is transferred from the dolicol to amide N of an asparagine residue of growing polypeptide chain. 4. trimming of the carbohydrate chain begins as the protein moves through the RER. 5. the golgi further trims and adds monosarride units
Exaplin in detail what occurs in the ER and the golgi
syntheis: protein part is snythesized in ribosomes, attached to RER. 1
1. N-linked- initially in the lumen of the ER and dolichol phosphate is required
2. Attachement of oligosaccrides
- O-Linked – In Golgi apparatus
- Incorporation of individual carbohydrate residues is catalyzed by specific glycosyl transferases
- UDP is the common nucleotide required for the incorporation of most of these carbohydrate residues
- Mannose & Fucose require GDP as carrier
- NANA (Sialic acid) is incorporated through CMP (as CMP-NANA)
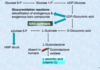
I-cell disease. What is it caused by? what does it lead to?
- Defects in the proper targeting of enzymes to the lysosomes
- Leads to the formation of dense inclusion bodies formation in the fibroblasts
Cause:
Deficiency in Phospho transferase to phosphorylate mannose residues in potential lysosomal enzymes
¯
Lack of mannose-6-phosphate tags in the enzymes, cannot reach the lysosomes
¯
Lysosomal enzymes secreted out of the cell, found in the plasma & urine
