Growth and puberty Flashcards
(17 cards)
Puberty
In boys

In males:
- Testicular enlargement to >4 ml volume measured using an orchidometer – the first sign of puberty
- Pubic hair growth – follows testicular enlargement, usually between 10 and 14 years of age
- Height spurt – when the testicular volume is 12–15 ml, after a delay of around 18 months.
- The height spurt in males occurs later and is of greater magnitude than in females, accounting for the greater final average height of males than females.
- In both sexes, there will be development of acne, axillary hair, body odour and mood changes.
- If puberty is abnormally early or late, it can be further

Growth and puberty

There are four phases of normal human growth:
-
Fetal
This is the fastest period of growth, accounting for about 30% of eventual height -
The infantile phase
Growth during infancy to around 18 months of age is also largely dependent on adequate nutrition. Good health and normal thyroid function are also necessary -
Childhood phase
This is a slow, steady but prolonged period of growth that contributes 40% of final height. Pituitary growth hormone (GH) secretion acting to produce insulin-like growth factor 1 (IGF-1) at the epiphyses is the main determinant of a child’s rate of growth, provided there is adequate nutrition and good health. -
Pubertal growth spurt
Sex hormones, mainly testosterone and oestradiol, cause the back to lengthen and boost GH secretion. This adds 15% to final height. The same sex steroids cause fusion of the epiphyseal growth plates and a cessation of growth

Puberty
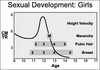
In girls

In females the features of puberty are:
- Breast development – the first sign, usually starting between 8.5 and 12.5 years
- Pubic hair growth and a rapid height spurt – occur almost immediately after breast development
- Menarche – occurs on average 2.5 years after the start of puberty and signals that growth is coming to an end, with only around 5 cm height gain remaining.
Short stature
Turners Syndrome

Short stature
Noonans syndrome

Short stature

- Short stature is usually defined as a height below the second centile (i.e. two standard deviations (SD) below the mean) or 0.4th centile (−2.6 SD).
- Only 1 in 50 children will be shorter than the 2nd centile and 1 in 250 (4 in 1000) shorter than the 0.4th centile

Short stature
Russell-Silver
is a congenital condition. It is characterized by stunted growth and limb or facial asymmetry. Symptoms range over a broad clinical spectrum from severe to so mild that they go undetected. The disorder is caused by very rare genetic defects.

Short stature
Downs syndrome

Tall stature

Familial:
Most common cause
Obesity:
Puberty is advanced, so final height centile less than in childhood
Secondary:
- Hyperthyroidism
- Excessive sex steroids - precocious puberty
- XS adrenal androgen steroids
- congenital adrenal hyperplasia
- True gigantism (xs GH secretory)
Syndromes:
- Long legged tall stature
- Marfan syndrome
- Homocystiuria
- Klinefelter syndrome
Proportionate tall stature at birth
Maternal diabetes
Primary hyperinsulinism
Beckworth syndrome
Sotos sydrome - large head, characteristic facial features and learning difficulties

Abnormal head growth

Causes of a large head:
- Tall stature
- Familial macrocephaly
- Raised intracranial pressure
- Hydrocephalus – progressive or arrested
- Chronic subdural haematoma
- Cerebral tumour
- Neurofibromatosis
- Cerebral gigantism (Sotos syndrome)
- CNS storage disorders, e.g. mucopolysaccharidosis (Hurler syndrome).
Precocious puberty

Precocious puberty (PP) may be categorised according to the levels of the pituitary-derived gonadotropins, follicle-stimulating hormone (FSH) and luteinising hormone (LH):
- Gonadotropin-dependent (central, ‘true’ PP) from premature activation of the hypothalamic–pituitary–gonadal axis
- Gonadotropin-independent (pseudo, ‘false’ PP) from excess sex steroids.

Precocious puberty
Females

Idiopathic or familial and follows the normal sequence of puberty. Organic causes are rare and are associated with:
- dissonance, when the sequence of pubertal changes is abnormal, e.g. isolated pubic hair with virilisation of the genitalia, suggesting excess androgens from either congenital adrenal hyperplasia or an androgen-secreting tumour
- rapid onset
- neurological symptoms and signs, e.g. neurofibromatosis.
Ultrasound examination of the ovaries and uterus is helpful in establishing the cause of precocious puberty. In the premature onset of normal puberty, multicystic ovaries and an enlarging uterus will be identified.

Precocious puberty
Males

This is uncommon and usually has an organic cause, particularly intracranial tumours. Examination of the testes may be helpful:
- Bilateral enlargement suggests gonadotropin release, usually from an intracranial lesion
- Small testes suggest an adrenal cause (e.g. a tumour or adrenal hyperplasia)
- A unilateral enlarged testis suggests a gonadal tumour.
- Tumours in the hypothalamic region are best investigated by cranial MRI scan

Delayed puberty
Causes of delayed puberty

Constitutional delay of growth and puberty/familial:
By far the commonest
Low gonadotropin secretion (hypogonadotropic hypogonadism)
- Systemic disease
- Cystic fibrosis, severe asthma, Crohn disease, organ failure, anorexia nervosa, starvation, excess physical training
- Hypothalamo-pituitary disorders
- Panhypopituitarism
- Isolated gonadotropin or growth hormone deficiency
- Intracranial tumours (including craniopharyngioma)
- Kallmann syndrome (LHRH deficiency and inability to smell)
- Acquired hypothyroidism
High gonadotropin secretion (hypergonadotropic hypogonadism)
- Chromosomal abnormalities
- Klinefelter syndrome (47 XXY)
- Turner syndrome (45 XO)
- Steroid hormone enzyme deficiencies
- Acquired gonadal damage
Post-surgery, chemotherapy, radiotherapy, trauma, torsion of the testis, autoimmune disorder.

Disorders of sexual differentiation

- The fetal gonad is initially bipotential. In the male, a testis-determining gene on the Y chromosome (SRY) is responsible for the differentiation of the gonad into a testis.
- The production of testosterone and its metabolite, dihydrotestosterone, results in the development of male genitalia. In the absence of SRY, the gonads become ovaries and the genitalia female.

Disorders of sexual differentiation
- The fetal gonad is initially bipotential. In the male, a testis-determining gene on the Y chromosome (SRY) is responsible for the differentiation of the gonad into a testis.
- The production of testosterone and its metabolite, dihydrotestosterone, results in the development of male genitalia.
- In the absence of SRY, the gonads become ovaries and the genitalia female
Congenital adrenal hyperplasia (CAH)

Congenital adrenal hyperplasia
Autosomal recessive disorder of adrenal steroid biosynthesis
Females present with virilisation of the external genitalia
Males present with salt loss (80%) or tall stature and precocious puberty (20%)
Long-term medical management with lifelong glucocorticoids, mineralocorticoids/sodium chloride if salt loss
Additional corticosteroids to cover illness or surgery
Salt-losing adrenal crisis needs urgent treatment with hydrocortisone, saline and glucose given intravenously
Monitor growth, skeletal maturity, plasma androgens and 17α-hydroxyprogesterone
Surgery for females.
- A number of autosomal recessive disorders of adrenal steroid biosynthesis result in congenital adrenal hyperplasia.
- Its incidence is about 1 in 5000 births, and it is commoner in the offspring of consanguineous marriages.
- Over 90% have a deficiency of the enzyme 21-hydroxylase, which is needed for cortisol biosynthesis.
- About 80% are also unable to produce aldosterone, leading to salt loss (low sodium and high potassium). In the fetus, the resulting cortisol deficiency stimulates the pituitary to produce adrenocorticotrophic hormone (ACTH), which drives overproduction of adrenal androgens.

