Hematología Clínica Flashcards
(179 cards)
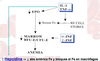
Hematopoyesis
Formación de los elementos sanguíneos en los órganos hematopoyéticos
Las células sanguíneas se generan a partir de…
célula madre
Para que se produzca la maduración celular es necesario:
- Micromedioambiente hematopoyético
- Citocinas que regulan la diferenciación celular
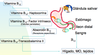
Evoluación de los órganos hematopoyéticos
Después de nacer se usa la MO. Al principio todos los huesos, peor conforme pasan los años se centra solo en los huesos planos

Esquema diferenciación de las líneas hematopoyéticas

Caracterísitcas de la célula madre hematoloyética
- Capacidad de autorrenovación, de proliferación y de diferenciación
- Plasticidad: Diferenciación hacia diversas líneas
- Inmunofenotípicamente son CD34+
Partes de la MO
- Contenido -> Células
- Ambiente / Estroma formado por:
- Fibroblastos
- Adipocitos
- Macrófagos
- Células endoteliales
- Células reticulares adventiciales
- Contienent -> Órgano hematopoyético
Funciones del estroma hematopoyético:
- Crear el µambiente necesario para que cada la diferenciación de cada célula progenitora
- El MEC dará soporte físico a las células progenitoras (las fija) mediante moléculas de adhesión, para que sobre ellas actúen citocinas producidas por las propias células del estroma
Tipos de citoquinas hematopoyéticas
1. Factores de crecimiento hematopoyético
- GM-CSF
- G-CSF
- M-SCF
- Eritropoyetina
- Trombopoyetina
2. Interleucinas-> IL1-12
- Genes que codifican estos factores
- Cromosoma 5: IL-3, -4, -5; GM-, M-CSF
- Cromosoma 7: EPO, IL-6
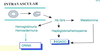
Regulación de la hematopoyesis

Qué implica necesariamente el síndrome anémico?
hipoxia tisular
Fisiopatología del síndrome anémico
- Curva disociación Hb a la derecha
- Redistribución vascular
- Aumento Gasto cardiaco
- Aumento EPO

De qué dependen las consecuencias clínics de la anemia?
- Intensidad
- Modo de instauración: Aguda (hVol - Shock) Crónica (hipoxia)
- Edad y estado de salud previo
- Capacidad de adaptación: mecanismos de adaptación a la anemia
Mecanismos de adaptación a la anemia:
- Intraeritrociatarios: hipoxia tisular -> 2-3 DPF
-
Extraeritrocitarios -> Redistribución sanguínea
- > > Ventilación
- > < actividad física - Aumento de la EPO
Sintomatología de la anemia
Síntomas dependientes de:
- Adaptación: taquicardia, disnea, soplos, cardiomegalia, taquipnea
- ↓ Hb -> Palidez cutaneo-mucosa (labios, conjuntiva y pliegues palmares)
-
↓O2 tisular ->
Generales: Astenia , irritabilidad, falta de concentración
Cardiacos. Angina pectoris. I. Cardiaca
Extremidades: Calambres musculares, claudicación intermitente - Cerebrales: Cefaleas, acúfenos, mareos
- Relacionadas con la enfermedad de base
Clasificación del sd anémico
- Según etiopatogenia: anemias reactivas y arreactivas
- Según VCM
Tipos de anemias regenerativas
Son las considereadas periféricas
- Pérdida de sangre aguda
- Anemias hemolíticas (corpusculares y extracorpusculares)
Tipos de anemias arregenerativas
Son las consideradas centrales
- ∆ de la célula madre
- Por desplazamiento
- Déficitis y/o trastornos metabólicos de factores eritropoyéticos
Anemias microcíticas
- Ferropenia
- Inflamatoria crónica
- Talasemia
- Sideroblástica
Anemias macrocíticas
- Hemolítica
- Déficit vitamínico (B12-fólico)
- SMD (Sd mielodisplásico?)
Anemia normocítica
- Anemia inflamatoria crónica
Metodología en el diagnóstico del síndrome anémico
- ¿Existe anemia?: Anamnesis y exploración física. Medir Hemoglobina
-
¿Cuáles son las características morfológicas?
Hematíes, Hb, hematocrito
VCM, HCM
Morfología frotis SP - **¿Cuál es el mecanismo de producción?: **Anamnesis, antecedentes, Hª familiar. Reticulocitos
- ¿Cuál es la causa fundamental?
- ¿Cuál es el tratamiento?= ULTIMO PASO
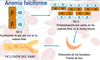
Definición de anemia ferropénica
Anemia hipocrómica y microcítica producida por déficit de Fe
Muy frecuente (afecta a 500 millones de peronas): paises en vías de desarrollo (80%) y en desarrollados (10-30%). Es una causa muy común de consulta médica (20% mujer fértil y ancianos y 10-15% en adolescentes)
Cómo y donde se absorve el Fe
En forma ferrosa en el yeyuno donde se transforma a forma férrica