Neurosurgery Board Review Flashcards
To rock the neurosurgery board examination. (162 cards)
0
Q
Alexander disease
A
- AD leukodystrophy on chrom 17
- GFAP protein problem
- Rosenthal fibers
- widespread demyelination in brain (frontal lobes involved first)
1
Q
Metachromatic leukodystrophy
A
- AR leukodystrophy
- sulfatase A
- increased cerebroside sulfate
- pathology -> PAS with macrophages
- Maroteaux-Lamy syndrome (MPS) has deficient sulfatase B
2
Q
Krabbe disease (globoid leukodystrophy)
A
- AR leukodystrophy on chrom 14
- galactocerebrosidase deficiency
- pathology shows globoid macrophages
- psychosine accumulates
- moderate/severe brain atrophy with firm white matter

3
Q
Tay-Sachs disease (GM2 gangliosidoses)
A
- AR on chrom 15 (lysosome)
- mutated hexosaminidase A (B is increased)
- cherry red macula
- also see Sandhoff’s disease (deficient B enzyme)
- gangliosides stain strongly for Luxol fast and Sudan black
- Balloon neurons on pathology
4
Q
Wolman Disease
A
- AR (lysosomes)
- deficient acid lipase -> accumulated lipids
- calcification of the adrenal gland may be seen
5
Q
Turner syndrome
A
- 45X
- short stature, no major CNS issues, aortic coarctation
6
Q
Miller-Deiker syndrome
A
- chromosome 17p (most cases are sporadic and not inherited)
- lissencephaly
7
Q
Batton Disease
A
- AR multiple genes
- most are due to thioesterase deficiency
- most common neuronal ceroid lipofuscinosis
8
Q
Prader-Willi Syndrome
A
- chromosome 15p
- MR, hypogonad, obese
9
Q
Pelizaeus-Merzbacher Syndrome
A
- X-LINKED leukodystrophy
- myelin proteolipid abnormality
- pathology -> segmental demyelination
10
Q
Adrenoleukodystrophy
A
- X-LINKED leukodystrophy
- ATP binding protein (ABCD1) peroxisome membrane transport protein
- pathology -> perivascular inflammation
- parieto-ccipital and deep white matter most affected
- spares subcortical U fibers -
- treatment is with Lorenzo’s oil
11
Q
Gaucher disease
A
- AR leukodystrophy
- deficient beta-glucocerebrosidase (increased glucosylceramide levels)
- pathology -> crumbled paper tissue cells
12
Q
Nieman-Pick disease
A
- AR leukodystrophy on chromosome 11
- type A/B 2/2 sphingomyelinase
- type C 2/2 cholesterol esterification
- cherry red macula
- Gaucher cells can be seen anywhere in the body, positive for PAS
13
Q
Farber Disease
A
- AR
- acid ceramidase deficiency
14
Q
Fabry disease
A
- X-LINKED
- deficient alpha-galactosidase (beta galactosidase in GM1 gangliosidoses)
- purple skin lesions and pain in hands/feet
15
Q
GM1 gangliosidosis
A
- AR
- deficient beta-galactosidase (increased levels of gangliosides, oligo and polysaccharides)
- increased keratin sulfate -> Morquio syndrome
- cherry red macula
17
Q
Kearns-Sayre Disease
A
- mitochondrial disease, +RRF, negative LA
- progressive external ophthalmoplegia
- heart block, retinitis pigmentosa
- DDX is myasthenia gravis which improves with cholinergics
- pathology -> spongiform cerebral and midbrain lesions
18
Q
MERRF (myoclonic epilepsy with ragged red fibers)
A
- mitochondrial DNA (8344)
- epilepsy and delay
- +RRF, NO lactic acidosis
18
Q
MELAS (myopathy, encephalopathy, lactic acidosis, and stroke)
A
- mitochondrial DNA
- +LA and +RRF (only mt dz that has BOTH)
- strokes that straddle vascular distributions (immediate subcortical white matter with hyperintense gyri on FLAIR/T2)
- MRS shows lactate doublet
19
Q
Cowden syndrome
A
- AD on chromosome 10
- PTEN loss of function (tumor suppressor)
- increased risk of many cancers
- CNS -> macrocephaly/hydrocephalus and dysplastic gangliocytoma of cerebellum (Lhermittos Duclos disease)
20
Q
Refsum disease
A
- AR (peroxisomes)
- deficient phytanoyl hydroxylase (increased phytanic acid = long chain FA)
- night blindness
- demyelinating sensory neuropathy
- hearing loss
- cardiomyopathy
21
Q
Gerstmann syndrome
A
- dominant parietal lobe (angular and supramarginal gyrus) insult
- symptoms: acalculia, agraphia, finger agnosia, left/right confusion
22
Q
Raeder’s Syndrome (paratrigeminal syndrome)
A
- lesions in and around carotid siphon
- trigeminal neuralgia (involving V1 and V2, NOT V3)
- Partial Horners (sweating is intact)
23
Q
Gradenigo’s Syndrome
A
- petrous apex lesions (otits, tumors, inflammation)
- retro-orbital pain
- 6th nerve palsy
- ear drainage if secondary to petrositis from ear infection
24
Vernet's Syndrome
- jugular foramen lesions
- CNs IX, X, XI
25
Villaret Syndrome
- posterior retroparotid space
- CNs IX, X, XI, XII, sympathetic chain
26
Tapia Syndrome
- posterior retroparotid space
- CNs X, XII (sometimes IX, VII, and sympathetic chain)
27
Schmidt Syndrome
- CNs X, XI
28
Garcin Syndrome
- skull base lesions (nasopharyngeal tumors and carcinomatous meningitis secondary to leukemia)
- all 12 cranial nerves
29
Collet-Sicard Syndrome
- Villaret syndrome WITHOUT sympathetic chain
- anterior occipital condyle lesions (tumors, vertebral aneurysms)
- CNs IX, X, XI, XII
30
Leber's Hereditary Optic Neuropathy
- mitochondrial DNA (3 known mutations)
- NEITHER lactic acidosis or RRF
- central visual loss bilaterally (not always complete) due to AV shunting in retina
31
Leigh Disease (Subacute Necrotizing Encephalomyelopathy)
- mitochondrial DNA and nuclear DNA (RANBP2 nuclear pore protein)
- + lactic acidosis
- spongy necrosis 2/2 myelin degeneration in the thalami, putamen, brainstem, and cord (hemispheres are spared)
32
Wyburn-Mason Syndrome
- multiple intracranial AVMs (mostly mesencephalic / optic pathway including retina)
- cutaneous nevi
- similar to Osler-Weber-Rendu syndrome
- not considered hereditary, congenital in origin
33
Osler-Weber-Rendu (hereditary hemorrhagic telangectasia)
- AD inheritance 2/2 numerous mutations (related to TGF and BMP signalling)
- multi-organ AVM formation
- cerebral abscesses / strokes 2/2 pulmonary AVMs
- many mucocutaneous telangiectasias
34
Tuberous sclerosis (Bourneville-Pringle disease)
- AD inheritance chrom 9 (hamartin) or 16 (tuberin)
- cortical tubers (ddx is Taylor focal cortical dysplasia, T1 dark, T2 bright gyrus)
- subependymal nodules -\> SEGAs in 15%
- white matter lesions extending radially from ventricles to cortex
- seizures (infantile spasms)
- cardiac rhabdomyomas, kidney angiomyolipomas/ADPCKD/RCC
- skin shagreen patches (lethargy regions), adenoma sebaceum, ash leaf spots
- peringual fibromas
35
Cri du Chat
- chromosome 5p
- cat-like cry
36
Trisomy 9
- Dandy-Walker malformation
- Subependymal and choroid plexus cysts
37
Patau Syndrome
- trisomy 13
- holoprosencephaly (no hemispheric division)
- retinoblastoma
- rocker-bottom feet and polydactyl
38
Edward's Syndrome
- trisomy 18
- gyrus dysplasia, cerebellar hypoplasia
- callosal agenesis
- Chiari II malformations
- scaphocephaly (dolichocephaly)
39
Dermatomyositis
- B-cell disease -\> myositis and vasculitis
- heliotrope rash and Gottron's papules (scaly macules on extensor surfaces of arms)
- 10% malignancy association
41
Canavan disease (aminoacidopathy)
- AR -\> aspartoacylase (unable to break down acetyl-aspartate to aspartate and acetate)
- white matter vaculoziation with Alzheimer's type II astrocytes (enlarged clear nuclei), does NOT spare subcortical U-fibers (in fact, preferentially demyleinates subcortical U-fibers)
- megaloencephaly
- abnormally long mitochondria

42
Wilson's disease (hepatolenticular degeneration)
- AR chromosome 13
- ATPase-7B protein (7A for Menkes) = cation transporter on mitochondria
- increased copper levels and decreased ceruloplasmin levels (both are decreased in Menkes)
- Alzheimer's type II astrocytes (large pale nuclei) -\> also seen in hepatic encephalopathy
- Opalski cells -\> microglia with eccentric prominent nuclei
- copper in putamen \> caudate \> BG/thalamus/cortex
- T1 and T2 show bright basal ganglia (hyperintense)
42
Von Gierkes (GSD type I)
- AR -\> glucose-6-phosphatase deficiency
- hepatomegaly
- severe hypoglycemia
43
McArdle's (GSD type V)
- AR -\> myophosphorylase deficiency
- most benign GSD
- renal failure 2/2 myoglobinuria
- increased serum CK
- excersize induced cramping
44
Pompe's (GSD type II)
- AR -\> acid-maltase deficiency
- infantile and juvenile/adult onsets
- early death for all forms 2/2 myopathy, cardiomegaly, and respiratory failure
45
Cori's (GSD type III)
- AR -\> amylo-1/6-glucosidase deficiency (debranching enzyme)
- hepatomegaly, seizures, growth retardation
46
Tauri's (GSD type VII)
- AR -\> phosphofructokinase deficiency
- symptoms are similar to McCardles (type V)
47
GSD type IX
- X-LINKED (only GSD that is X-linked) -\> phosphoglycerate kinase deficiency
- symptoms different than other GSDs: hemolytic anemia, MR, seizures/tremor
48
Sialidosis (mucolipidoses type I)
- alpha-neuramidase deficiency
- cherry red macula
- macroglossia
- coarse facial features
49
Sandhoff syndrome (GM2 gangliosidoses)
- AR chromosome 5
- hexosaminidase B gene deficiency (A accumulates)
- in Tay-Sachs (A mutation, and B accumulates)
50
Crouzon Syndrome (acrocephalosyndactyl type II)
- AD chromosome 10
- FGF-receptor 2 gene
- most common craniosynostosis syndrome
- NORMAL intelligence
51
Apert syndrome (acrocephalosyndactyl type I)
- AD chromosome 10 (FGF-receptor 2 gene)
- bicoronal craniosynostosis (most common) -\> brachycephaly or turricephaly
- Harlequin orbit (elevation / elongation of superolateral orbit on craniosynostotic side)
- MENTAL RETARDATION (different than Crouzon)
- intracranial abnormalities in 50% (callosal dysgenesis, hydrocephalus, cavum veragae)
- cervical spine issues
52
Saethre-Chotzen Syndrome (acrocephalosyndactyl type III)
- AD chromosome 7 (TWIST gene)
- "cotton beaten" skull xrays
- coronal synostosis most common
- syndactyl between 2nd-3rd digits (Carpenter syndrome is 3rd-4th digits)
- normal intelligence
54
Waardenburg syndrome (acrocephalosyndactyl type IV)
- AD \>\> AR (multiple genes involved - SOX, PAX, etc)
- depigmentation syndrome (skin, eyes, hair)
- hearing loss common
- cerebellar/cortial hypoplasia
- peripheral nerve demyelination -\> Hirschsprung's disease
55
Pfeiffer syndrome (acrocephalosyndactyl type V)
- AD (FGF receptors 1 AND 2)
- several subtypes (normal intelligence in type I)
- multi-suture synostosis (kleeblattschadel)
- brachydactyl (also thumb/big toe point away from other digits)
56
Carpenter syndrome
- AR (RAB23 gene -\> vesicle trafficing protein, ONLY AR synostosis syndrome)
- syndactyl (3rd/4th digits)
- crypthorchidism in all males
- heart abnormalities (dextrocardia)
56
MEN type I (Wermer syndrome)
- AD -\> MEN1 gene
- pituitary adenomas (only MEN with pit adenomas)
- pancreatic tumors (gastrinoma most common)
- lipoma / angiolipoma
- parathyroid hyperplasia
57
Greig syndrome
- AD (GLI3 gene on chromosome 7)
- metopic and/or sagittal synostosis
- poly and syndactylyl
- callosal dysgenesis and ventriculomegaly
58
MEN type IIa (Sipple syndrome)
- AD chromosome 10 (RET gene)
- pheochromocytoma (33%)
- medullary thyroid cancer (100%)
- parathyroid hyperplasia
59
MEN type IIb
- RET gene
- pheochromocytoma (50%)
- medullary thyroid cancer (85%)
- marfanoid
- mucosal neuromas (100%)
60
Turcot syndrome
- AD
- type I = hereditary non-polyposis colorectal CA -\> DNA mismatch mutations -\> GI/GU tumors + astrocytomas
- type II = familial adenomatous polyposis -\> APC gene -\> GI tumors + medulloblastomas + craniofacial exostosis
61
von-Hippel-Lindau syndrome
- AD on chromsome 3 - increased erythropoeitin, PDGF, VEGF, TGF - hemangioblastomas of brain and retina - endolymphatic sac tumors of posterior petrous bone - clear cell RCC (type I and type IIb) - pheochromocytomas (type II VHL)
62
Basal cell nevus syndrome (Gorlitz syndrome)
- AD chromosome 9 - PTCH gene - Tumors: basal cell carcinomas, odontogenic tumors (80%), medulloblastoma (4 to 25%) - dural calcification along falx and tentorium - skeletal anomalis (ie: bifid ribs)
63
Schwannomatosis
- sporadic mutations in SMARCB1 (IN11/hSNF5) -\> chormatic remodeling protein - multiple schwannomas of spine/equina/cutaneous/and nonvestibular CNs
64
Li-Fraumeni syndrome
- AD - TP53 mutations - CNS disease: astrocytomas, choroid plexus carcinoma, and PNETS - Body disease: breast CA, osteosarcomas, many other tumors can occur
65
Meningioangiomatosis
- hamartomatous lesion of meningiothelial cells, fibroblasts - involves pia and underlying cortex - frequently calcified - DDX is malignant/invasive meningioma
66
Neurocutaneous melanosis
- excessive proliferation of melanocytes - leptomeningial invasion (bright of T1) - melanocytic deposits seen in amygdala, cerebellum, pons, thalami, inferior frontal lobes - associated with Dandy-Walker (10%) - malignant melanoma occurs in 40 to 60%
68
Haberland syndrome (encephalocraniocutaneous lipomatosis)
- nevus psiloliporus -\> fatty lipoma of scalp (underlies an area of alopecia usually) - posterior fossa lipomas (bright on T1, dark on CT) - spinal lipomas
69
Menkes kinky hair disease
- X-LINKED - ATPase-7A transporter protein (7B in Wilson's) - LOW levels of copper AND ceruloplasmin (HIGH levels in Wilson's) - neuropsychiatric issues - myelin of cortex/cerebellum affected - tortuous intracranial vessels (elastic fiber issues)
70
Hallervoden-Spatz disease (pantothenate kinase associated neuropathy)
- AR chrom 20 (PANK2 gene)
- iron deposition in medial GP and substantia nigra reticulata
- dementia, spasticity, movement problems
- tauopathy with neurofibrillary tangles
- spares cortex (different than other Fe diseases)
- hypointense GPe on T2 images (sometimes bright middle = "eye of the tiger" sign)
71
Neuroferritinopathy
- AD chrom 19 (ferritin light chain protein) - iron deposition in cortical and BG neurons - symptoms around 40 yo (extrapyramidal s/s) - NO cognitive issues (different than many other disorders like Huntington's that have similar presentations)
72
Infantile neuroaxonal dystrophy
- AR PLA2G6 gene (phospholipase A2 protein) - cerebellar atrophy - iron deposition in GP/SN in 50% - progressive tetraparesis - also spares cortex (like Hallervoden-Spatz)
73
Aceruloplasminemia
- AR chrom 3 - putamenal iron (NOT copper) - diabetes, retinopathy, extrapyramidal, dementia - affects cortex, cerebellum, and BG
74
Dejerine-Sota disease (hereditary motor and sensory neuropathy type III)
- both AD and AR forms - hypertrophic nerves - sensori-motor demyelination (NCS \< 12 m/s = severe) - increased CSF protein (unlike other HMSNs)
75
Charcot-Marie-Tooth type I
- AD chrom 17 (peripheral myelin protein) - sensori-motor demyelination - late childhood/early adolescence -\> difficulty running, atrophy of feet and hand muscles -\> pes cavus, hammertoes, high arches
76
Charcot-Marie-Tooth type II
- AD - sensori-motor axonal neuropathy (demyel in type I) - decreased CMAP amplitudes - same onset and findings as type I (difficulty running, pes cavus, hammertoes, high arches)
77
Phenylketonuria
- AR chrom 12 phenyalanine hydroxylase - also can be caused by deficient tetrahydrobiopterin reductase enzyme - phenylalanine cannot be broken down into tyroxine, which is also needed to make melanin - blond hair / blue eyes (no melanin formation) - musty urine odor (phenylacetate) - MR and seizures
78
Maple syrup urine disease (leucinosis)
- AR chrom 19 (all components of the branched chain keto-dehydrogenase complex/protein) - increased levels of leucine, isoleucine, valine (all are branched, essential AAs) - sotolone in urine is diagnostic (smells like caramel) - spongy white matter lesions
79
Homocystinuria
- AR chrom 21 cystathione synthase protein - unable to break down homocysteine into cystathione - B6 vitamin (pyridoxine) is co-factor for cystathione synthase protein - homocysteine can be converted to methionine via methionine synthase (B12 (cobalamin) as cofactor), but this also requires intact folate (vitamin B9) cycle - Clinically: marfanoid, codfish vertebrae (biconcave), strokes (2/2 intimal fibrosis), anterior lens dislocations
80
Hartnup disease
- AR chrom 5 AA transporter protein in kidney/gut - unable to absorb tryptophan, which is needed to make niacin (B3), melatonin, and serotonin - mostly asymptomatic - pellagra like symptoms can develop: dementia, diarrhea, dermatitis - aminoaciduria
81
Hyper-glycine-emia (glycine encephalopathy)
- AR mutations in proteins that encode the glycine decarboxylase complex/enzyme (GCDS gene is most commonly mutated) - callosal agenesis and white matter vacuoles
82
Urea acid cycle disorders
- X-LINKED ornithine carbamoyl-transferase deficiency (most common cycle disorder) - increased ammonia levels - increased orotic acid in urine - type II Alzheimer cells are seen frequenly (also seen in Wilson's disease)
83
Canavan's disease
- AR
- aspartoacylase deficiency -\> increased acetyl-aspartate -\> interference with myelin production
- pathology -\> spongy white matter
- clinically -\> seizures, blind, hypotonia
84
Sturge-Weber syndrome
- sporadic inheritance
- facial port wine stain in distribution of V1/V2
- gliosis and calcification of ipsilateral brain -\> seizures and neuro deficits
- leptomeningial and ocular angiomas
85
Hunter syndrome (MPS type II)
- X LINKED recessive
- deficient iduronate sulfatase
- NO corneal issues (compared to Hurler's syndrome)
86
Duchenne muscular dystrophy
- X LINKED recessive
- increased lumbar lordosis and scoliosis
87
Osler-Weber-Rendu (hereditary hemorrhage telangiectasia)
- AD
- multi-organ AVMs
- pulmonary AVMs -\> paradoxial stroke / cerebral abscess formation
88
Tuberous sclerosis
- AD (chrom 9 -\> hamartin protein, chrom 16 -\> tuberin protein), although many are de-novo mutations
- seizures start as infantile spasms progress to Lennox-Gestaut
- EEG -\> hypsarrhythmic ("mountainous" rhythm) that is INTER-ictal
- CNS -\> cortical tubers (with balloon type cells), subependymal giant cell astrocytomas (+ for neuron enolase, synaptophysin, neurofilament, +/- GFAP), subependymal nodules (can be calcified)
89
ARDS (acute respiratory distress syndrome)
- Bilateral chest infiltrates
- PaO2 : FiO2 \< 200 (if \< 300 then acute lung injury is present)
- acute onset
- normal PACWP (normal left atrial function)
90
Infantile spasms (West syndrome)
- associated with tuberous sclerosis
- EEG -\> hypsarrhythmia (INTER-ictal high voltage mountainous rhythm)
- seizures frequently progress to Lennox-Gastaut type around 4 to 5 years of age
- treatment -\> ACTH
91
Juvenile myoclonic epilepsy
- myoclonic jerks that can progress to GTC seizures (usually occur in morning)
- normal intelligence
- EEG -\> 4-6 Hz spke/wave discharges with polyspikes
- treatment -\> valproic acid
92
Petite mal (abscence)
- Blank staring spells
- EEG -\> 3 Hz spike/wave discharges (inter-ictal EEG is normal)
- treatment -\> ethosuximide
93
Lennox-Gastaut
- refractory epilepsy and mental retardation
- EEG -\> 1-2 Hz spike/waves
- treatment -\> valproic acid (usually requires multi-drug treatments)
94
Benign focal epilepsy of childhood
- GTCs at night, focal seizures during day, normal intelligence
- usually 5 to 9 year olds
- EEG -\> di/tri phasic sharp waves over Rolandix cortex
- treatment -\> carbamazepine (usually a self limited condition that ends in adulthood)
- AD inheritance (usually a family history)
95
Pulmonary embolism EKG findings
- sinus tachycardia (most common)
- "S1, Q3, T3" -\> wide S complex in V1, large Q wave in V3, and inverted T wave in V3 (not sensitive)
- inverted T waves in V1, V2, and V3
- right axis deviation
- right bundle branch block
96
Right and left bundle branch blocks
- WiLLiaM MaRRoW (M pattern in V1-V2 and W pattern in V3-6 for RBBB, opposite for left)
97
Foix-Alajouanine syndrome
- progressive LE neurologic symptoms 2/2 type I spinal AV fistula
- weakness, B/B/sexual dysfunction, back pain
98
Pallister-Hall syndrome
- chromosome 7p -\> GLI3 gene
- associated with hypothalamic hamartomas -\> precocious puberty, gelastic seizures, and behavior problems
- other body malformations (digits, epiglottis, larynx, cardiac, renal, anal)
99
Carney complex
- AD (protein kinase A subunit gene)
- GH pituitary adenomas in 10%
- adrenal adenomas -\> ACTH independent Cushing's disease in 33-50%
- other endocrine tumors
- myxomas (cardiac)
- spotty skin pigmentation
- schwannomas (melanotic variant)
100
Annulus tendineus (of Zinn)
- part of SOF
- all rectus muscles
- nerves: CN II, superior and inferior division of III, CN VI, nasociliary branch of V1 (frontal branch of V1 does NOT)
- ophthalmic artery (NOT vein)
101
McCune-Albright disease
- GNAS1 mutations (same in other fibrous dysplasia lesions)
- endocrine abnormalities (most commonly precocious puberty)
- cafe-au-lait spots
- fibrous dysplasia (polyostotic)
102
Mazabraud syndrome
- multiple muscular myxomas
- polyostotic fibrous dysplasia
- middle aged women
103
Chordoma (pathology\_
- notochord remnants
- S100 +, cytokeratin + (chondrosarcomas are neg for cytokeratin)
- sacrum \> clivus \> vertebral bodies
- worse prognosis than chondrosarcoma

104
endothelial proliferation
- associated with high grade gliomas IV
- contrast enhancement

105
desmoplastic medulloblastoma
- SHH mutations
- better prognosis
- more often located off midline
- reticulin stain is what differentiates these from other medulloblastomas

106
granulomatous inflammation
- classically seen in TB
- giant multi-nucleated Langerhan's cells may be seen

107
pleomorphic xanthoastrocytoma
- substantial nuclear pleomorphism
- large cells with "foamy" cytoplasm are a common clue

108
AT/RT tumors
- usually \< 3 years old
- eccentric nuclei with intracytoplasmic inclusions (helps distinguish from medulloblastoma)
- positive EMA
- negative BAF47 protein staining (2/2 chromosome 22q11.2 deletion of hSNF1/INI1 gene), BAF47 is + in medulloblastoma

109
Primary CNS lymphoma (pathology)
- most commonly diffuse large B cell (approximately 3% of all primary brain tumors)
- CD20, CD19, CD79a + (normal T-cells will stain for CD3, but very small number)

110
Dejerine and Roussy syndrome
- infarcts of VPL and VPM
- thalamic pain
- decreased sensation
- transient hemiparesis
- abnormal movements
111
Lewy body (pathology)
- Parkinson's disease
- many other Parkinson-like syndromes
- a-synuclein, ubiquitin, neurofilaments, beta crystallin
- pale halo sign
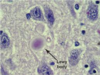
112
Pick bodies (pathology)
- tau, ubiquitin, tubulin
- cytoplasmic, "hug" nuclei, well demarcated
- associated with dementias

113
Lafora bodies (pathology)
- associated with Lafora progressive myoclonic epilepsy
- can be seen anywhere in the body, in the CNS affect neurons (not astrocytes)
- polysaccharide inclusions (polyglucosans)
- cytoplasmic

114
Bunina bodies (pathology)
- associated with ALS
- cystatin C and transferrin protein
- intracytoplasmic, very small in size

115
Hirano bodies (pathology)
- seen in Alzheimer's and Pick's dementia
- actin protein
- cytoplasmic, located mainly in CA1 of hippocampus

116
Zebra bodies (pathology)
- seen in gangliosidoses and Hurler's syndrome
- appear like a mitochondria with stripes

117
Negri bodies (pathology)
- seen in Rabies
- affect Purkinje neurons
- cytoplasmic
- appear like a red blood cell trapped in a cell
118
Germinomas
- round neoplastic cells with clear cytoplasm
- inflammatory infiltrate distinguishes these from other germ cell tumors
- pineal \> suprasellar location (may have synchronous lesions)
- hyperintense on T1/T2, avidly enhance
- placental alkaline phosphatase specific, CD117 is sensitive marker

119
Meningioma (pathology)
- + for EMA and vimentin
- calcium in 25% of cases
- grade III (papillary, rhabdoid, anaplastic)
- grade II (chordoid, clear cell, atypical)
- monosmy of chromosome 22 most common genetic mutation (NF2 is allelic loss of 22q)
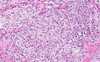
120
Metastatic carcinoma (pathology)
- distinct margin from surrounding brain

121
Paragangliomas
- arise from extra-adrenal chromaffin cells (analogous to pheo, but do not normally secrete catecholamines)
- filum terminale \>\> middle ear (glomus tumor)
- Zellballen chief cells -\> + synaptophysin, + chromogranin
- smaller number of sustentacular cells -\> + S100
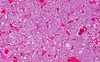
122
Oligodendroglioma (pathology)
- fried egg appearance only seen on final (not frozen section)
- deletion of chrom 1p 19q
- isocitrate dehydrogenase mutations are also common

123
Langerhan's cell histiocytosis
- thickened pituitary stalk (DI)
- antigen presenting cells
- Birbeck granules on electron microscopy
- lytic skull lesions
124
Dressing apraxia
- posterior right parietal lobe injury
125
Writing apraxia
- left angular gyrus injury
126
Prosopagnosia
- medial inferior temporo-occipital injury
127
Astereogenesis
- Injury to either parietal lobe
128
Seesaw nystagmus
- posterior diencephalon / pretectum
- para/suprasellar lesions
- lateral medulla
129
Spasmus mutans
- triad of head nodding, nystagmus, and head turning in infants
- etiology is unclear
130
Brun's nystagmus
- pontomedullary junction
- vestibular pathways
- seen in 11% of patients with acoustic neuromas
- when looking to lesion the nystagmus is slow and high amplitude, when looking away fast and low amplitude
131
Ocular bobbing
- central pontine lesions
132
Ocular myoclonus (opsoclonus-myoclonus)
- lesions in Guillain-Mollaret's triangle (ipsilateral inferior olive/red nucleus, and contralateral dentate nucleus)
- also seen in neuroblastoma
133
Upbeat nystagmus
- medulla
- ventral tegmentum
- cerebellar pathways (usually anterior vermis)
134
Downbeat nystagmus
- cervicomedullary junction
- can be seen in patient's with Chiari malformation
135
Ocular flutter
- opsoclonus-myoclonus (ocular myoclonus) in horizontal plain ONLY
- cerebellar pathways and brainstem
136
Convergence-retraction nystagmus
- Parinaud syndrome of dorsal midbrain / pretectum
137
I-cell disease (mucolipidoses type II)
- autosomal recessive
- phosphotransferase deficiency (unable to add phosphate groups to mannose residues in golgi apparatus)
- corneal clouding, developmental/physical delay
- resembles Hurler syndrome
138
Hurler syndrome (MPS type 1)
- autosomal recessive on chromosome 4
- iduronidase enzyme
- corneal clouding, developmental/physical delay
139
Sanfillipo syndrome (MPS type III)
- autosomal recessive on multiple chromosomes (8, 12, and 17)
- multiple enzymes
- type A -\> sulfamidase (heparan sulfate N-sulfatase)
- type B -\> N-acetylglucosamidase
- developmental delay and behavioral problems (in addition to features of the MPS syndromes = coarse facial features, hirsutism, delay), no corneal clouding, urinary heparan sulfate excretion in type B
140
Morquio syndrome (MPS IV)
- autosomal recessive on multiple chromosomes (3, 16)
- galactose-6-sulfatase (type A) or beta-galactosidase (type B) deficiencies
- level of keratan sulfate increased (urinary excretion)
- severe skeletal dysplasias, dwarfism, cardiac/respiratory issues may cause death
- few neurologic problems
141
Maroteaux-Lamy syndrome (MPS VI)
- autosomal recessive
- deficient sulfatase B (aka: N-acetylgalactosamine-4-sulfatase), metachromatic leukodystrophy also has deficient aryl-sulfatase
- normal intelligence
- severe skeletal problems (kyphoscoliosis)
- thickening of dura and corneal clouding
- treated with galsulfase (Naglyzme) one of the most expensive drugs in the world
142
Meckel-Gruber syndrome
- autosomal recessive on multiple chromosomes
- encephalocele, microcephaly, microophthalmia, cleft lip/palate, polydactyly, polycystic kidneys, heart disease, ambiguous genitalia, pulmonary hypoplasia
- maternal hyperthermia on days 20 to 26
143
Essential tremor
- overactivity in olives
- AD inheritance
- 6 to 8 Hz
- type of postural tremor that is pathologic
144
Multiple sclerosis
- associated with DR2, A3, B7, and DR15 alleles
- predominately T-cell
- oil red O stain shows active de-myelination
- myelin destruction via macrophages
- acute MS (Marburg variant) -\> rapidly fatal in children
- acute MS (Balo's disease) -\> rapidly progressive, prominent central necrosis with large plaques
145
Acute disseminated encephalomyelitis (ADEM)
- T-cells
- occurs after viral/vaccine
- 80% make full recovery
- peri-VENULAR inflammation
146
Progressive multifocal leukoencephalopathy (PML)
- reactivation of papovirus (JC virus) in immunocompromised
- asymmetric demyelination, mostly deep white matter
- oligodendrocytes with viral inclusions surround white matter loss
147
Marchiafava-Bignami disease
- demyleination of corpus collosum (medial aspects)
- Italian men who drink cheap red wine
- death within 3-6 years of symptom onset
148
SEGA (pathology)
- tuberous sclerosis
- near foramen of monro
- + synaptophysin, + neuron enolase, + neurofilament, +/- GFAP

149
Langerhan's cell histiocytosis
- 3 separate diseases based on severity, usually affects children
- eosinophilic granuloma (most benign, local bony involvement, spine/skull)
- Hand-Schiller-Christina disease (bone and visceral involvement)
- Letterer-Siwe disease (fatal in early childhood)
150
Birbeck granules (pathology)
- seen in Langerhan's cell histiocytosis
- electron microscopy

151
Aneurysmal bone cyst (pathology)
- lytic/expansile/cystic
- blood products
- giant cells
- "egg shell" cortex

152
Aneurysmal bone cyst (imaging)
- lumbar \> thoracic \> cervical
- arch \> body

153
Osteiod osteoma (pathology)
- "osteoblastoma" if \> 1.5cm
- can cause "painful" scoliosis (most common cause of painful scoliosis in children -\> resection cures scoliosis if done within 15 months)
- tx with aspirin
- central "nidus" interlacing osteoid with loose vascular stroma
154
Osteoid osteoma (imaging)
- central nidus with surrounding sclerosis
- sharply demarcted from surrounding bone
- posterior elements \> body
- lumbar spine most common location
- technitium bone scans show intense activity on intermediate and delayed films
- CT is best (often missed on plain films or MRI)

155
Osteochondroma (pathology)
- cartilage "cap" on cortical bone
- continguous with parent bone near lesion
- sometimes part of hereditary multiple osteochondromas (increased risk of chondrosarcoma)
- long bones affected most

156
Common radiation tolerances
- cord up to 50 Gy (usually given in 2 Gy fractions)
- chiasm 9 to 10 Gy
- radiation necrosis seen more commonly with total doses larger than 50 Gy
157
Septo-optic dysplasia (Morsier syndrome)
- two key features
(a) absent septum pellucidi
(b) optic nerve/chiasm hypoplasia
- also associated with other abnormalities
(a) olfactory bulb abscence (bulbs are present in lobar HPE)
(b) heterotopias/polymicrogyria/schizencephaly
158
tCreutzfeldt-Jacob disease
- "vaculozation" throughout brain
- protein 14-3-3 in CSF
- biphasic or triphasic sharp waves on EEG

159
Zellweger syndrome (cerebrohepatorenal syndrome)
- spectrum that includes Refsum's disease and adrenoleukodystrophy
- AR inheritance in many genes regarding peroxisome function
- increased amount of fatty acids (do not get broken down)
160
Gitelman syndrome
- AR defect in thiazide sensitive sodium/chloride co-transporter
- distal convulted tubule
- **hypokalemia / hypochloremic metabolic ALKALOSIS**
- hypomagnesemia / hypocalcemia may also be present
161
Bartter syndrome
- multiple genetic mutations
- ion transporters located on thick ascending loop of Henle (similar to being on a loop diuretic all the time)
**- hypokalemic / hypochloremic metabolic ALKALOSIS**
- dehydration / hypotension with elevated renin / aldosterone levels
162
Dexamethasone suppression test
- **no suppression** of cortisol secretion in adrenal adenomas (ie: low ACTH levels and high cortisol with no suppression because adenoma functions autonomously)
- **no suppresion** of cortisol secretion in tumors that ectopically secrete ACTH (high ACTH levels, high cortisol levels, ectopic tumor cells do not have feed back inhibition and therefore continue to secrete ACTH even in presence of decadron)
- **suppression of cortisol** secretion with ACTH **pituitary adenomas** with **high dose test**, but not low dose (high ACTH levels, but pituitary retain feedback inhibition and therefore cortisol level is decreased with decadron)
