Pulm/Renal - Biochemistry - The Golgi Apparatus: Mucolipidoses; Glycobiology: Mucopolysaccharidoses & Sphingolipidoses Flashcards
(204 cards)
Describe the hypothesized signalling mechanism by which proteins that are being synthesized end up in the rough endoplasmic reticulum.
(1) Translation begins in the cytosol; an RER-targeting sequence on the partially synthesized protein is bound by a signal recognition particle (SRP).
(2) Upon binding to an SRP receptor in the RER, GTP hydrolysis opens an associated translocon protein (the RER channel opens, and the SRP dissociates from the ribosome).
(3) Translation continues directly through the translocon and into the RER lumen; signal peptidase cleaves the signalling sequence on the synthesized protein.

In the signal hypothesis of RER synthesis, what binds to signalling sequences on partially synthesized proteins?
Where are these proteins when this occurs?
Where are they taken next?
Signal receptor particles (SRPs);
the cytosol;
SRP receptors on the RER

Secretory protein synthesis begins in the cytosol. How are these proteins targeted to the RER?
What effect does this targeting have on translation?
By signal receptor proteins (SRPs) that bind SRP receptors on the RER surface;
translation is blocked when SRPs bind the partially synthesized proteins, but it is restored when the SRP dissociates and the ribosome is bound to the SRP receptor

What must occur for both signal receptor proteins to dissociate from SRP receptors on the RER and also the associated translocons to open?
GTP hydrolysis

Secretory proteins begin their translation in the ________, where an N’ signalling sequence targets them towards the RER.
Upon entering the RER, what cleaves this signalling sequence from the newly synthesized proteins?
Cytosol;
signal peptidase (in the RER membrane)

Signal receptor particles (SRPs) bind what to bring partially synthesized secretory proteins to the RER?
An N’ signalling sequence on the partially synthesized protein and the 60s ribosomal subunit

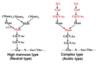
What are the two main purposes of protein glycosylation?
What are some other purposes?
Folding, targeting;
E-leaflet protein stability, enhanced cell-to-cell contact
________ are carbohydrate-binding proteins, and can interact with other glycosylated proteins.
Lectins
Lectins are ___________-binding proteins, and can interact with other ___________ proteins.
Carbohydrate;
glycosylated
True/False.
Glycosylation is typically the addition of single glucose monomers to amino acid side chains.
False;
it is usually the addition of glycans (either single oligosaccharides or complexes) to amino acid side chains.
What are the two types of protein glycosylation?
O-linked;
N-linked
N-linked glycosylation occurs in the _____________.
O-linked glycosylation occurs in the _____________.
RER;
Golgi apparatus
Describe O-linked glycosylation (in terms of starting substrate and any involved amino acids or enzymes).
Give an example.
Individual carbohydrates are sequentially added to –OH groups of serine or threonine by glycosyl transferases;
the formation of ABO blood antigens
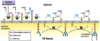
Describe N-linked glycosylation (in terms of starting substrate and any involved amino acids or enzymes).
What amino acid sequence is required?
Preformed oligosaccharide complexes are added to asparagine residues as the peptide is translocated through the RER membrane (translocation process shown in attached image);

Asn - X - (Ser/Thr)
__-linked glycosylation is characterized by the addition of preformed oligosaccharide complexes to __________ residues.
__-linked glycosylation is characterized by the sequential addition of individual carbohydrates to ___________ residues.
N, Asparagine (Asn-X(Ser/Thr);
O, Serine/Threonine
Describe the steps of synthesis of the oligosaccharide complex used in N-linked glycosylation.
(1) UDP-linked sugars are added to dolichol phosphate from the cytosolic side.
(2) This complex then ‘flips’ from the cyotosolic to the RER lumen side.
(3) The completed oligosaccharide complex can then be cleaved and added to asparagine residues by oligosaccharyl transferase.

In synthesis of the oligosaccharide complexes used in N-linked glycosylation, _______-linked sugars are first added to _____________ _____________ on the cytosolic side of the RER membrane.
UDP;

dolichol phosphate
What must occur for the oligosaccharide complexes used in N-linked glycosylation to enter the RER lumen?
It ‘flips’ from the cytosolic side to the RER lumen

What is the first substrate for synthesis of the oligosaccharide complexes involved in N-linked glycosylation?
It is attached to what substance in the RER membrane?
What substance can be used in the lab to inhibit this process, thus disrupting proper protein folding?
UDP-GluNAc;
dolichol phosphate;
tunicamycin

N-linked glycosylation is accomplished via what enzyme?
Oligosaccharyl transferase

What effect does tunicamycin have on cells?
What is its clinical use?
Inhibition of first step of N-linked glycosylation
–> inhibition of proper protein folding in eukaryotic cells;
none (laboratory use only)
True/False.
Disulfide bonds form in many proteins synthesized both in the cytosol and the RER.
False.
Disulfide bonds form mainly in secretory proteins and proteins of the E-leaflet;
they are only made in RER-synthesized proteins
Why do disulfide bonds only form in the RER lumen?
Via what enzyme?
It is an oxidative environment;
protein disulfide isomerase
What is the most common genetic cause of liver disease (jaundice, cirrhosis) in children?
What is the most common disorder necessitating pediatric liver transplants?
α-1 antitrypsin deficiency;
α-1 antitrypsin deficiency