Week 1 Flashcards
(125 cards)
What are the sites of haematopoesis (production of blood cells) at the following points in development?
- Embryo
- Birth
- Birth to maturity
- Adult
Embryo - yolk sac, then liver, then marrow. 3rd-7th month - spleen
Birth - mostly bone marrow, liver and spleen when needed
Birth to maturity - number of active sites in bone marrow decreases but ability for haematopoiesis is retained
Adult - bone marrow of skull, ribs, sternum, pelvis and proximal ends of femur
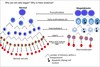
Describe the haemopoietic tree
Long-term haematopoietic stem cells (self-renewing) become short term haematopoietic stem cells, which then become multipotent progenitor (MPP) cells
These then differentiate into either common myeloid progenitor (CMP) cells or common lymphoid progenitor (CLP) cells
CMP
- megakaryocyte-erythrocyte progenitors (MEP)
- erythrocytes
- platelets
- granulocyte-monocyte progenitors (GMP)
- granulocytes
- macrophages
- Pro-DC
- dendritic cells
CLP
- Pro-DC
- dendritic cells
- Pro-T
- T cells
- Pro-NK
- NK cells
- Pro-B
- B cells

Describe the steps from the Common Myeloid Progenitor (CMP) cell to the mature neutrophil (granulopoiesis)
CMP > myeloblast
Myeloblast > promyelocyte
Promyelocyte > myelocyte
Myelocyte > metamyelocyte > N. band forms >…
Neutrophil

Describe the steps from the Common Myeloid Progenitor (CMP) cell to mature red cell/erythrocyte (erythropoiesis)
What changes occur in the cell morphology, and at what point?
CMP > pronormoblast
Pronormoblast > basophilic/early normoblast (ribosome synthesis)
basophilic/early normoblast > polychromatophilic/intermediate normoblast (haemoglobin accumulation, nucleus condenses)
polychromatophilic/intermediate normoblast > orthochromatic/late normoblast
Nucleus is then lost, some RNA retained
Reticulocyte > Mature red cell/erythrocyte (remaining RNA is completely lost over ~7 day period)

Describe the steps from the Common Myeloid Progenitor (CMP) to platelet
CMP > megakaryoblast
Megakaryoblast > promegakaryocyte
Promegakaryocyte > megakaryocyte
Platelets form on the periphery of the megakaryocyte and bud off
What are the types of granulocyte?
Eosinophils
Basophils
Neutrophils
Describe the structure and function of neutrophils
Structure
- Segmented (polymorphic) nucleus
- Stains neutrally (hence the name)
Functions
- short life in circulation (transit into tissues)
- phagocytoses invading molecules
- kills invaders with granule contents, killing itself in the process
- in doing so, attracts other cells
- numbers are increased by body stress e.g. infection, trauma etc.
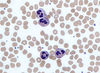
Describe the structure and function of Eosinophils
Structure
- bi-lobed (usually)
- bright orange/red granules
Function
- fight parasitic infections
- involved in hypersensitivity reactions and often elevated in patients with allergic conditions

Describe the structure and function of Basophils
Structure
- infrequent in circulation
- large, deep purple granules, obscuring the nucleus
Function
- circulating version of tissue mast cell
- mediates hypersensitivity reactions
- Fc Receptors bind IgE
- granules contain histamine

Describe the structure and function of monocytes (macrophages)
Structure
- large, singular nucleus
- faintly staining granules
Function
- circulate for a week, then enter tissues to become macrophages
- phagocytose invaders, killing them and presenting antigens to lymphocytes
- attract other cells
- more long-lived than neutrophils
How does a lymphocyte change in appearance when it becomes activated (a.k.a. from mature to atypical)?
Mature - small, with condensed nucleus and a rim of cytoplasm (B)
Activated - large, with plentiful blue cytoplasm extending around neighouring red cells (A)

Effector cells are easily identifiable, but how are the precursors identified?
Immunophenotyping - examine the expression profile of proteins (antigens) on the surface of the cells. Done with mAbs w/ fluorescent tags
What is a common site for bone marrow aspiration?
Posterior iliac crests
Describe some of the key features of a typical red blood cell
Full of Hb
No nucleus - can’t divide or replace damaged proteins meaning lifespan is limited (approx 120 days)
No mitochondria - limited to glycolysis for energy generation
Flexible - requires specialised membrane
High surface area/volume ratio
What structures in the membrane of a RBC allow it to be flexible (like a hiking tent!)?
Protein ‘spars’ - alpha and beta spectrin
Describe the structure and function of haemoglobin
Structure
- Tetrameric globular protein made up of 2 alpha and 2 beta chains (in adults)
- Haem group Fe2+ is found in the centre of this protein in a flat porphyrin ring
Function
- Oxygen delivery
- Acts as a buffer for H+
- Involved in CO2 transport
Describe the process of RBC destruction
Usually occurs in the spleen
Old RBCs are taken out of the circulation by macrophages and red cell contents are recycled
- globin chains are broken down into amino acids
- haem group is broken down into iron and bilirubin
Briefly describe the regulation of RBC production
Hypoxia sensed by kidneys
Erythropoietin is produced which stimulates RBC production in the bone marrow
More RBCs are released, and EPO levels subsequently drop
How does a RBC a) generate ATP and b) prevent Fe2+ from becoming Fe3+ (oxidation)?
ATP is generated via glycolysis or Embden-Meyerhof pathway which yields a net generation of ATP and NADH
NADH prevents the oxidation of Fe2+ to Fe3+

What protects our RBCs from reactive oxygen species such as hydrogen peroxide?
What is the rate limiting enzyme in the recycling of this molecule?
Glutathione (GSH) reacts with hydrogen peroxide to form water and an oxidised glutathione product (GSSG). This is then replenished by NADPH.
The rate-limiting enzyme in this process is glucose-6-phosphate dehydrogenase (G6PD), a deficiency of which can potentially result in anaemia in individuals that are unable to compensate by producing more reticulocytes e.g. in conditions of high oxidative stress, such as infection

How is CO2 transported to the lungs?
60% transported as bicarbonate (RBCs are important in generation)
30% transported bound directly to haemoglobin - carbamino-Hb
10% is dissolved in solution
Describe the dissociation curve for oxygen and why it is unusual
Unusual as it doesn’t follow Michaelis-Menten kinetics. Dissociation curve is sigmoidal
As one O2 molecule binds to a subunit, the Hb shape changes (allosteric effect) to increase its affinity for binding further O2 molecules
What does a shift to either the right or the left in the oxygen dissociation curve mean?
What factors may cause this to happen?
Right shift indicates decreased oxygen affinity of Hb allowing more oxygen to be available to tissues
Left shift indicates increased oxygen affinity of Hb, allowing less oxygen to be available to tissues
Left Shift
- increased pH
- decreased CO2
- decreased temperature
- decreased 2,3-DPG
- foetal haemoglobin
- myoglobin
Right Shift (Bohr Effect)
- decreased pH
- increased CO2
- increased temperature
- increased 2,3-DPG
Anaemia is defined as a reduced total red cell mass, however this is not easy to measure in routine practice. What two surrogate markers are used to assess anaemia, and how are they measured?
Haemoglobin concentration - spectrophotometer
Haematocrit - ratio of whole blood that is red cells if a sample was allowed to settle, normally about 50%