1 - 100 Flashcards
(100 cards)
A 16 month-old girl presents with patchy alopecia, whorled erythematous scaly eruption, and asymmetric limb shortening. What laboratory or radiologic test may aid in diagnosis?
1 Brain MRI
2 Alkaline phosphatase
3 Chest radiograph
4 Bone films
5 Complete blood count
Bone films
The patient has Conradi-Hunermann Syndrome. This is a X-linked dominant disorder characterized by ichthyosiform erythroderma in Blaschko’s lines in infancy which resolves with follicular atrophoderma, patchy alopecia, short stature, cataracts, scoliosis, assymetric limb shortening. Bone films will demonstrate stippled epiphyses. Ichthyosis and stippled epiphyses resolve after infancy.
A patient with 20 nail dystrophy, steatocystoma multiplex and natal teeth likely has a mutation in the genes coding for:
1 Keratins 5
2 Laminin 5
3 Plakophilin 1
4 Keratins 6b & 17
5 Keratins 6 &16
Keratins 6b & 17
Pachyonychia congenital is an autosomal dominant condition with 20 nail dystrophy. The patient described has Type II (Jackson-Sertole) disease, which includes steatocystoma multiplex, natal teeth, multiple cysts, and micropthalmia, and is caused by mutations in keratins 6b& 17. Type I (Jadassohn-Lewandowsky) also includes focal symmetric PPK, follicular hyperkeratosis, oral leukokeratoses and is caused by mutations in keratins 6 &16. Type III includes the clinical features of type I + corneal leukokeratosis. Mutations in keratins 5&14 represents EB simplex, Laminin 5 mutation is seen in Junctional EB, and plakophilin 1 mutation is seen in ectodermal dysplasia with skin fragility.
Which of the following is defective in Ehlers-Danlos syndrome (EDS) with congenital adrenal hyperplasia?
1 Tenascin-X
2 Lysyl oxidase
3 Lysyl hydroxylase
4 None of these answers are correct
5 All of these answers are correct
Tenascin-X
Tenascin-X defects are associated with EDS and with congenital adrenal hyperplasia. The phenotype is that of typical EDS with hyperextensible skin, hypermobile joints, and tissue fragility. Lysyl oxidase is defective in X-linked EDS (type V) and Occipital horn syndrome (type IX). Lysyl hydroxylase is defective in ocular-scoliotic (type VI) EDS.
Which type of porphyria is associated with hyponatremia?
1 Acute intermittent porphyria
2 Porphyria cutanea tarda
3 Variegate porphyria
4 Hereditary coproporphyria
5 Erythropoietic protoporphyria
Acute intermittent porphyria
Acute intermittent porphyria can cause hyponatremia due to the syndrome of inappropriate antidiuretic hormone secretion.
Hypoplasia of the breast can be seen in which disease?
1 Anhidrotic ectodermal dysplasia
2 Maffucci syndrome
3 Congenital syphilis
4 Marfan syndrome
5 Osteogenesis imperfecta
Anhidrotic ectodermal dysplasia
Anhidrotic ectodermal dysplasia is a X-linked recessive disease caused by mutations in ectodysplasin, a member of the tumor necrosis family. Patients may have dry skin with pigmentation periorbitally, hypohidrosis, sparse hair, hypo-anodontia, nail dystrophy, and frontal bossing, and saddle nose deformity. In addition to abnormalities of other ectodermally derived structures, the breast and nipple-areolar complex may be absent or hypoplastic.
Which disease is found more commonly in mothers of patients with chronic granulomatous disease?
1 Sarcoidosis
2 Erythema nodosum
3 Churg-Straus disease
4 Wegener’s disease
5 Discoid lupus erythematous
Discoid lupus erythematous
Female carriers of chronic granulomatous disease have an increase incidence of discoid lupus, infections and apthous stomatitis.
A patient presents with several light blue cyst-like lesions on the eyelid. They consult their list of problems and bring up plantar hyperkeratosis and dysplastic toenails. On oral exam, you note that they have both upper and lower dentures. The patient relates that after losing their “baby teeth”, only 3 teeth grew in their place. What syndrome does this person most likely have?
1 Schopf-Schulz-Passarge
2 Gardner syndrome
3 Hypohidrotic ectodermal dysplasia
4 Cowden syndrome
5 Cronkhite-Canada
Schopf-Schulz-Passarge
Schopf-Schulz-Passarge syndrome is associated with hydrocystomas of the eyelids, hypotrichosis (near complete loss of hair early in life), hypodontia, nail abnormalities and multiple palmoplantar eccrine syringofibroadenomas. The other listed syndromes do not fit the description above.
A child presenting with the scalp findings shown and a right arm hypoplasia would be diagnosed with which of the following syndromes?
1 Adams-Oliver syndrome
2 Bart’s syndrome
3 Progeria
4 Dunnigan syndrome
5 None of these options are correct
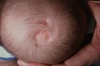
Adams-Oliver syndrome
Adams-Oliver syndrome is defined by aplasia cutis congenita (ACC) (shown in the image), usually of the midline scalp with limb hypoplasia. Bart’s syndrome also has ACC as a finding, but it is usually present on the lower extremities and associated dominant dystrophic epidermolysis bullosa. Progeria is a premature aging syndrome associated with a lamin-A mutation. Dunnigan syndrome is also known as Familial partial lipodystrophy and is associated with a mutation in the BSCL2 gene. Neither are associated with findings of ACC.
Which of the following findings is characteristic of a mutation in lamin A?
1 Lipoatrophic sclerodermoid skin
2 Alopecia
3 Craniomegaly with small face
4 Severe premature atherosclerosis with early death
5 All of the answers are correct
All of the answers are correct
A mutation in Lamin A causes Progeria (Hutchinson-Gilford syndrome). Other findings include nail atrophy and muscle/bone wasting. Presentation is in the first or second year of life. An increased urine hyaluronic acid can be helpful in diagnosis.
What is the mode of transmission for lamellar ichthyosis?
1 Autosomal dominant
2 Autosomal recessive
3 X-linked dominant
4 X-linked recessive
5 Sporadic

Autosomal recessive
Lamellar ichthyosis which is characterized by collodian membrane in newborns and platelike scale in children and adults is an autosomal recessive syndrome. The gene defect is transglutaminase 1 (TGM1).
Think Transglutaminase Fetal Rat
The most common cutaneous association with monilethrix is:
1 Eczema
2 Hypopigmentation
3 Hyperpigmentation
4 Keratosis Pilaris
5 Atrophy

Keratosis Pilaris
Monilethrix is an autosomal dominant condition which, by definition, presents with “beaded” hear. Clinically, patients present with short, sparse lusterless hair. Keratosis pilaris is the most common associated feature.

Which type of epidermolysis bullosa simplex is associated with early death?
1 Weber-Cockayne
2 Generalized (Koebner)
3 Dowling-Maera
4 Ogna variant
5 Non-Herlitz variant
Dowling-Maera
The Dowling-Maera variant of epidermolysis bullosa simplex is associated with widespread bullae, significant mucous membrane and laryngeal/esophageal involvement, nail dystrophy, and early death.
The arylsulfatase C gene is mutated in which disease?
1 X-linked ichthyosis
2 Refsum syndrome
3 Haim-Munk syndrome
4 Naxos syndrome
5 Griscelli syndrome
X-linked ichthyosis
Arylsulfatase C is also known as steroid sulfatase and is mutated in X-linked ichthyosis. This condition is inherited in a X-linked recessive pattern. Clinical findings include: brown scale sparing palms, soles and flexures, comma-shaped corneal opacities, failure of labor progression and cryptorchidism. It is also mutated in X-linked recessive type chondrodysplasia punctata.
Premalignant leukoplakia of the oral mucosa is associated with:
1 Bloom syndrome
2 Werner Syndrome
3 Xeroderma Pigmentosum
4 Dyskeratosis Congenita
5 Rothmund-Thomson syndrome
Q/Q(M)-474043 Report a Problem

Dyskeratosis Congenita Dyskeratosis Congenita (also known as Zinsser-Engman-Cole syndrome) is thought to have two modes of inheritance. The more common X-linked disorder is due to a mutation in the Dyskerin gene, while the autosomal dominant form is due to a mutation in TERC, a telomerase RNA component. Clinical features include reticulated gray-brown hyperpigmentation, paloplantar hyperkeratosis, alopecia, onychodystrophy, premalignant leukoplakia of any mucosal surface, and mental retardation.

Which cutaneous finding is seen in patients with phenylketonuria?
1 Angular stomatitis
2 Ichthyosis
3 Pigment dilution of hair and skin
4 Phyrnoderma
5 Erosive diaper dermatitis
What is the enzyme defect?
The accumulation of ally
What should the PKU patients avoid eating in their diet?

Pigment dilution of hair and skin
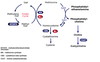
Phenylketonuria is an autosomal RECESSIVE condition caused by a mutation in the gene coding for phenylalanine hydroxylase**. Defect in this enzyme results in accumulation of phenylalanine and its metabolites. I_ncreased phenylalanine_** has toxic effects on the central nervous system in addition to competitively inhibiting tyrosine in melanogenesis. Inhibition of melanogenesis results in pigmentary dilution of the hair and skin. Other features of this condition include a predisposition to eczema, sclerodermoid changes of the skin, urine that has a distinctive “mousy” odor, psychomotor delay, mental retardation, seizures and hyperreflexia. A low-phenylalanine diet instituted early on can prevent these manifestations of the disease. The morbidity of phenylketonuria has improved since the advent of routine neonatal screening for this condition.
Phenylalanine ——-> Tyrosine —-> Melanin
+Phenylanine Hydroxylase
Phenyl Ketonuria (PKU) is a common metabolic disorder in growing children. A child suffering from PKU has defective legs and is usually unable to stand. Such a child may also be mentally retarded. This condition is due to a defect in the amino acid metabolism. In the course of amino acid metabolism there is a step in which the amino acid phenyl-alanine gets converted into another amino acid called tyrosine. This conversion requires specific enzyme called phenyl pyruvic hydroxylase. In the child suffering from PKU this particular enzyme will be absent and as a result phenyl-alanine gets converted into a ketone body phenyl pyruvic acid. It starts circulating throughout the body through the blood resulting in the disorder. The absence of the enzyme phenyl pyruvic hydroxylase is due to the expression of a recessive autosomal gene in the homozygous condition.
PKU can be detected by a simple urine test. Such children are usually treated with a diet, which is free from the amino acid phenyl-alanine.
Which syndrome is characterized by hyperhidrosis, lack of pain sensation, hypersalivation, and absent fungiform papillae?
1 Turner Syndrome
2 Noonan Syndrome
3 Riley-Day
4 Rubinstein-Taybi syndrome
5 Cornelia de lange Syndrome

Riley-Day
“Riley Day the IRON CHEF cutting his fingers”
Riley-Day syndrome is also known as Familial Dysautonomia. It is an autosomal RECESSIVE disorder with the gene defect on the long arm of chromosome 9.
Patients have unmyelinated sensory and sympathetic neurons and autonomic dysfunction, leading to hyperhidrosis, decreased corneal sensation and tear flow, hypersalivation, gastroesophageal reflux, decreased deep tendon reflexes, and lack of pain sensation.
They also exhibit abnormal histamine skin test.
Retention of primary teeth a dental finding of which of the following conditions?
1 Hypomelanosis of Ito
2 Letterer-Siwe disease
3 Tuberous sclerosis
4 Jackson Sertoli syndrome
5 Hyper-IgE syndrome

Hyper-IgE syndrome
Hyper-Immunoglobulin E syndrome is an autosomal dominant condition with impaired regulation of IgE function and deficient neutrophil chemotaxis. There is increased susceptibilty to infections and increased IgE serum levels. Retained primary teeth and lack of development of secondary teeth are characteristic findings. The remaining conditions do not have this as a prominent finding.

All of the following disorders are exacerbated by UV radiation and have the eye findings in photo except:
1 Bloom syndrome
2 Hartnup’s disease
3 Refsum syndrome
4 Cockayne syndrome
5 Rothmund-Thomopson syndrome

Refsum syndrome
Refsum’s syndrome is an autosomal recessive disorder caused by mutations in phytanoyl-CoA hydroxylase. Clinically, patients have mild icthyosis, cerebellar ataxia, polyneuropathy, salt and pepper retinitis pigmentosa, sensorineural deafness, and arrhythmias with heart block. They are not overly sensitive to UV radiation.

Homocystinuria is caused by a defect in:
1 Phenylalanine hydroxylase
2 Biotinidase
3 Holocarboxylase synthetase
4 Cystathione beta-synthetase
5 Gp91-phox
What is the most common eye abnormailty?
Cystathione beta-synthetase
Cystathione beta-synthetase is defective in homocystinuria, an autosomal recessive conditions characterized by increased homocystine and methionine levels in blood and urine. Other findings include a malar flush, DVTs/emboli, cardiovascular disease, livedo reticularis, leg ulcers, blonde hair/fiar complexion, downward lens dislocation, glaucoma, mental retardation, seizures, psychiatric disorders and a marfanoid body habitus. The other enzymes are not involved in this condition.
Cutaneous osteomas are seen in which syndrome?
1 Waardenburg syndrome
2 LEOPARD syndrome
3 Carney complex
4 Albright hereditary osteodystrophy
5 Gaucher�s syndrome
Albright hereditary osteodystrophy is caused by mutations in the Gs subunit of adenylate cyclase. There is calcification and ossification due to pseudohypoparathyroidism, absent 4th knuckle, and hypogonadism.
Q/Q(M)-474257 Report a Problem
A child with phenylketonuria likely presents with which cutaneous problems?
1 Blue-gray generalized hyperpigmentation
2 Alopecia universalis
3 Generalized hypopigmentation
4 Generalized hyperpigmentation
5 Leg ulcers
Phenylketonuria is an autsomal recessive disorder caused by a mutation on the long arm of chromosome 12. A deficiency of phenylalanine hydroxylase or its cofactor tetrahydrobiopterin leads to accumulation of phenylalanine. Clinical features include generalized hypopigmentation, eczematous dermatitis, sclerodermoid changes, seizures, psychomotor delay, urine with �mousy� odor, mental retardation.
Q/Q(M)-474030 Report a Problem
Patients with junctional epidermolysis bullosa have been found to have mutations in:
1 Laminin 5
2 Bullous pemphigoid antigen 2
3 Collagen 17
4 BP180
5 All of the answers are correct
All of the answers are correct. Laminin 5 is a protein integral in the adhesion of the dermis to the epidermis. Also involved in junctional epidermolysis bullosa is bullous pemphigoid antigen 2, collagen 17 and BP180, which are synonymous for the same structure.
Q/Q(M)-477712 Report a Problem
Which genetic defect could explain cutaneous findings in addition to abnormal immunoglobulin levels, recurrent respiratory infections, hypogonadism, and an increased risk of leukemia and lymphoma?
1 RecQL3
2 ERCC6
3 WAS gene
4 NADPH oxidase
5 Adenosine deaminase

Bloom’s syndrome is an autosomal recessive disorder caused by mutations in the RecQL3 gene encoding a DNA helicase. Clinically, individuals with Bloom’s syndrome have a photodistributed erythema with telangectasia on the malar eminences.
The may also have decreased IgM and IgA levels, hypogonadism**, and an increased risk for **leukemia and lymphoma.

What is the underlying gene defect for this transgrediens form of palmoplanter keratoderma
1 SLURP-1
2 TOC gene
3 Plakoglobin
4 Keratin type 1
5 Keratin type 9

Attached picture is Mal de Meleda (keratosis palmoplantaris transgrediens) which is an autosomal recessive form of diffuse PPK, associated with keratotic plaques that extend to the dorsal aspects of the hands and feet (“transgrediens”) and may overlie joints . Hyperhidrosis, superinfection, and occasionally perioral erythema, brachydactyly, and nail abnormalities are associated. Mal de Meleda is due to mutations in ARSB, which encodes SLURP-1. The other choices represent gene defects for “non-transgrediens” forms of PPK (Plakoglobin in Naxos syndrome, TOC gene in Howel-Evans syndrome, K1 in non-epidermolytic PPK “Unna-Thost”, and K9 in epidermolytic PPK “Vorner”