clinical and biochemical features of metabolic bone disease Flashcards
(72 cards)
what is a metabolic bone disease
a group of diseases that causes a change in bone density and bone strength
by indreaing resorption or decreasing bone formation or altering bone structure
it may be associated with disturbances in mineral mechanism
what are the 5 common metabolic bone disorders *
primary hyperthyroidism
rickets/osteomalacia
osteoporosis
pagets disease
renal osteodystrophy
what are metabolic symptoms of bone diseases
hyper/ocalcaemia
hyper/ophosphtaemia
what are the bone symptoms of metabolic bone disorders
deformity
fracture
what is hydroxyapatite made of
calcium and phosphorus
how is cancellous bone metabolically active
remodelling - 5% body is remodelling at any time
whole skeleton remodels over 7years
acts as a ca reserve - there is a balance with blood and bone
what makes a bone strong
mass - genetic
material properties - collagen cross-linking, woven/lamella, mineralisation (young bone less mineralised so ductile, older is more brittle), microcracks
microarchitecture - trabecular thickness, trabecular connectivity, cortical porosity (holes in corticies - when teenager, grow fast and fracture)
macroarchitecture - hip axis length and diameter
summarise the age related changes in bone mass *
build bone up until 20s,
stable until 40
lose
accelaratd loss in women in menopause - lose 30% of bone mass
both men and women have a slow phase

how can exercise change bone mass
change in dimention - add perisoteum
change in shape - lay down bone specific to force
describe bone remodelling during the growth of the tibea
lay bone down in anterior and posterior compartments - specific to minimise the mass of bone that is layed down
therefore there is increased bending strength anterio-posteriorly, than medio-laterally
bone is layed down in the periosteum region and resorbed from the endosteum
descrieb teh sexual dismorphism in bone growth
men develop bigger bones with a bigger cavity
women’s bones have thicker endosteum so are stronger, as they get older - endocortical bone resorption and periosteal bone formation occurs - never matches mens

descrieb cortical bone microfractures *
the bone has a structure to absorb energy - cortex has osteons and alternating density of lamellae
irreversible plastic deformation does occur - causing microfractures that dissipate the energy - they crack the matrix - usually limited to the interstitial bone between osteons - if they accumulate the bone strength will be comprimised
this is detected so has to be repaired
they are repaired through bone remodelling
each oseton represents a previous remodelling event

describe the bone remodelling cycle *
activation phase - osteocytes detect a microcrack - the crack crosses canaliculi so severing the osteocyte’s dendritic processes
this causes osteocytic apoptosis
this acts as a signal to the connected surface lining cells which are of osteoblast lineage
the lining cells and osteoblasts release factors that attract cells from the blood and marrow into the remodelling compartment, these cells are monocytes and
resorption phase - osteoclasts are generated locally from the recruited cells - they resorb the matrix and the offending microcrack
reversal phase - switch from resorption to formation, osteoclasts are apoptosed
formation - then osteoblasts deposit new lamellae bone
the osteoblasts that are trapped in the matrix become osteocytes, others die or form new osteoblast lining
osteoblasts last months, clasts last weeks so have to keep recruiting clasts
this is happening all over the skeleton

what are the biochemical investigations in bine disease *
serum
- bone profile - ca, corrected ca, phosphate, alkaline phosphate, mg
renal func
- creatinine
- PTH
- 25-hydroxy vut D
urine
- ca/phos
- NTX (bone resorptoion marker)
summarise Ca balance
some absorption from GIT but not very efficient
reabsorb some from kidney - but excrete some, this cannot be helped
there is a big influx in and out of bone - cancellous bone has a huge supply
how do you correct serum ca measurements
total ca is 2.15-2.56mmol/L
46% of this is protein bound
47% is fress ionised
7% is complexed
measure ionised in casualty
acid base balance affects ca - in alkalosis ca bind to protein = ca drop = tinglking feeling
venous stasis might falsely elevate levels
corrected ca = [ca] +0.02(45-[albumin])
if albumin is high, true ca is low - corrected ca compensates for the protein level
explain how PTH regulates ca levels *
has the predominant role in min by min ca reg
if ca drops PTH increases in minutes
immediately causes bone resorption by stimulating osteoclasts - releases ca and phos
acts on kidney - increase ca resorption in DCT, increase phos excretion by inhibiting NAP co-transporter in PCT (phosphaturic hormone), increases 1a hydroxylation so increases vit D activation = increase in intestinal ca and phos absorption
clinical relevance of PTH response system
84aa peptide but N1-34 is active - this is used clinically
mg dependant - alcoholics have low mg = low ca
PTH half life - 8mins - so can be measured intraoperatively
PTH receptor activated by PTHrP during breast feeding - releases ca from bone, PTHrP also produced by tumours so hypocalcaemia might be a presenting feature of a tumour
describe the relationship between PTH and ca *
a steep inverse sigmoidal function relates PTH and Ca
minimum - even at high levels there is always some PTH production
set-point - point of half max suppression of PTH, steep part of slope, small perturbation causes large change in PTH

describe the mechanism by which PTH drives ca reabsorption *
PTH increases the number and activity of TRPV5/6 (ca channels)on the lumen - increase ca reabsroption
also increases the active transport of ca into the blood out of the cells

describe how PTH causes bone resorption through the RANK system *
increases RANKL production = increase in osteoclast differentaition
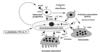
how common is primary hyperparathyroidism *
occurs in 50s
female:male 3:1
2% people develop it post-menopausally
what are the causes of primary hyperparathyroidism *
mainly parathyroid adenoma - benign
parathyroid hyperplasia - genetic condition when under 40
parathyroid carcinoma <1%
rare familial syndromes - MEN1, MEN2a HPT-JT
how do you diagnose primary hyperparathyroidism *
an elevated total/ionised ca with PTH levels frankly elevated or in the upper half of normal range (this is inappropriately high)
corrected ca >2.6mmol/l with PTH>3.9pmol/l (normal range 1-6.8)