What is complement?
Where are the components of complement synthesized?
“Complement” refers to a group of interacting plasma proteins that act collectively to mediate important biological activities that help defend against extracellular pathogens. The term is derived from the observations that these proteins helped, or “complemented” antibody in the destruction of bacteria by immune serum.
Synthesized mainly in liver, but local tissue synthesis by macrophages and other cells also is important
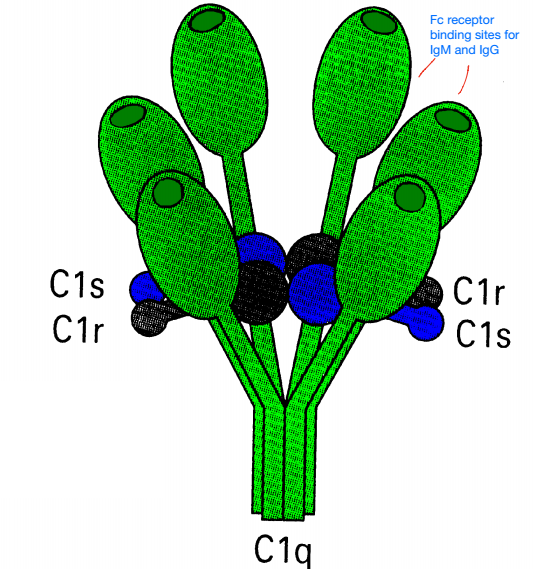
How many binding sites does C1 have? What does C1 bind?
How many subunits is C1 comprised of? What are the functions of the subunits?
What ion is required to maintain the C1 complex?
- C1 is composed of three subunits (subcomponents), named C1q, C1r, and C1s.
- C1q has 6 binding sites for Fc region of IgG, IgM. C1 has no enzymatic activity!
- C1r & C1s are serine proteases
- Ca++ is required to maintain complex
Initiation. When IgM or IgG1, IgG2, or IgG3 bind to an antigen, their rotational freedom is restricted in such a way as to expose a previously unavailable site on the Fc portion of the antibody which can bind to C1q. Since the C1 complex can thus recognize bound antibody, C1 is considered to be the recognition unit of the complement system.
The C1 subunits are held together by Ca++. Removal of calcium by chelators, such as EDTA or EGTA causes the complex to dissociate and prevents activation. C1q binds to antibody, and this induces a conformational change in C1r, allowing it to act enzymatically on both itself and C1s, producing the active enzyme from of C1s. The structure of C1 is shown below.
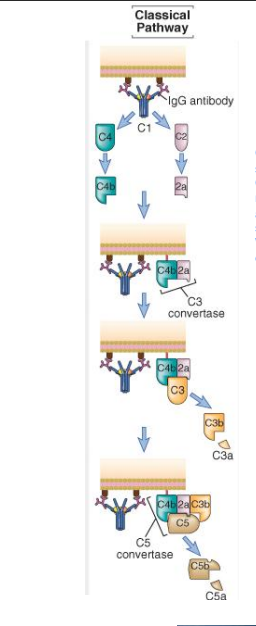
Explain the steps of the classical pathway.
The C1 complex binds either one IgM or two IgGs. The complex becomes activated and cleaves C4 into C4a and C4b. C4a floats off in solution and C4b binds covalently to the surface of the antigen. C1 also cleaves C2 into C2a and C2b. C2b floats off in solution and C2a binds non-covalently with C4b. C4b and C2a together form C3 convertase. C3 convertase is cabable of cleaving multiple C3 molecules. C3 convertase cleaves C3 into C3a and C3b. C3a floats off in solution while C3b binds covalenlty to the surface of the antigen. C3b molecules that bind close to C4b and C2a (C3 convertase) form a complex called C5 convertase which cleaves C5 into C5a and C5b.
attached is slide 6 of PP
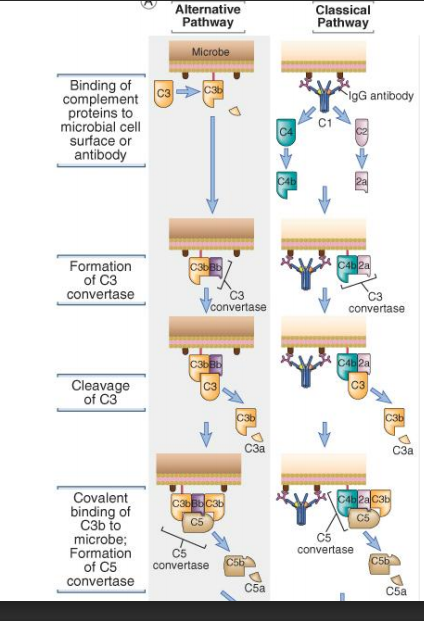
What is the alternative pathway also known as? What is the major difference between the alternative and classical pathways?
This additional pathway (alternative pathway) is sometimes called the properdin pathway, since this unique serum protein plays an important regulatory role. The major difference between the alternative and classical complement pathways is that the alternative pathway may be activated by antigens without the requirement for specific antibodies, thereby providing the biological activities so important to host defense in the critical early stages of infection, before the appearance of significant amounts of antibody.
Explain the sequence of the alternative pathway.
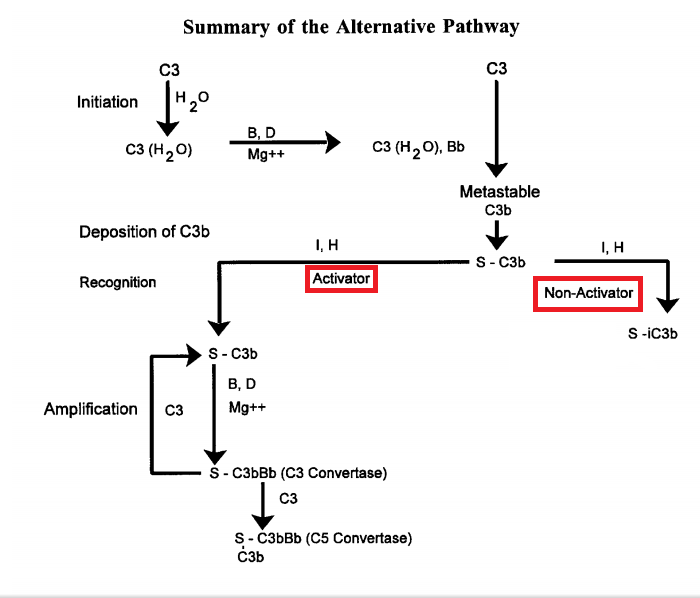
C3 is spontaneously hydrolyzed into C3a and C3b (note that this is a slow process). C3b binds covalently to the surface of the antigen. D (with Mg2+) cleaves B into Bb and Ba. Bb binds to C3 forming a C3 convertase (C3bBb). This enzyme is extremely labile, but the attachment of a molecule of properidn stabilizes it—> C3bBbP. The stable enzyme then cleaves more C3, forming more C3a and C3b.
Since many molecules of C3 may be activated this way, this is an important amplification point in the pathway. Of course each one of these C3b molecules is capable of generating new C3bBbP enzyme. Thus this reaction sequence is called the positive feedback loop of the alternative pathway.
The third multiunit enzyme of the alternative pathway is the C5 convertase. This is formed by the association of at least one additional molecule of C3b to the C3bdependent C3 convertase–>
(C3b)2BbP. This is capable of cleaving C5 into C5a and C5b.
please see pgs 111-112 of course notes
attached is slide 9 of PP
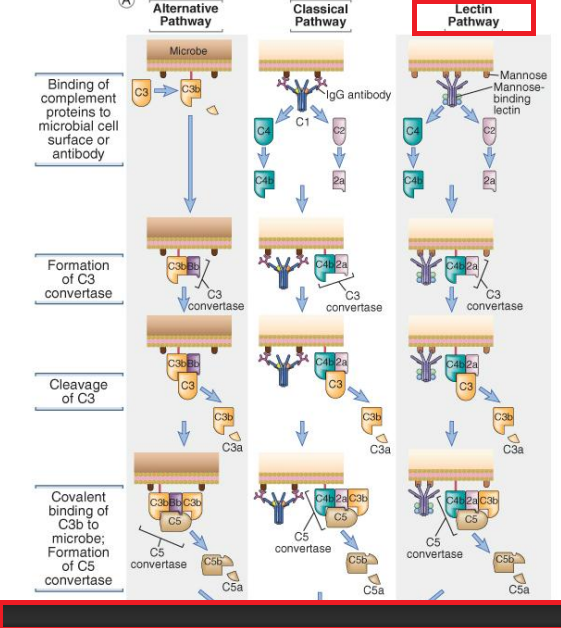
Explain the lectin pathway.
Mannose binding lectin (MBL) binds to the surface of the antigen (bacteria). Note that this binding is antibody independent. MBL, like C1q, it circulates in a complex with two associated serine proteases bound to it. These enzymes are called MBL-Associated Serine Protease 1 (MASP 1) and MBL-Associated Serine Protease 2 (MASP 2), and seem to serve the same roles as C1r and C1s in the classical pathway. MASP 1 and 2 cleave C4 and C2. The rest of the pathway proceeeds like the classical pathway.
C4b+C2a=C3 convertase
C3b binds covalently to the surface of the antigen. Multiple C3b molecules are made.
C4b+C2a+C3b=C5 convertase=C5 is cleaved into C5a and C5b
What is the common terminal pathway of the classical, alternative, and lectin pathways? What complement proteins are involved?
What organisms are destroyed via this pathway?
What specific antigen is this pathway critical for the destruction of?
Lysis: The C5b6789 complex (C5b-9) assembles on a membrane, inserts as a channel, and produces a disruption of the target cell’s ability to regulate solute and ionic gradients. The target may swell and burst, or just lose internal proteins causing death. Gram positive organisms, as well as fungi, and protozoal parasites are resistant to this lytic effect, so practically speaking, only some gram negative bacteria can be killed by C5b-9. Host defense against Neisseria species often requires lysis as a major protective mechanism.
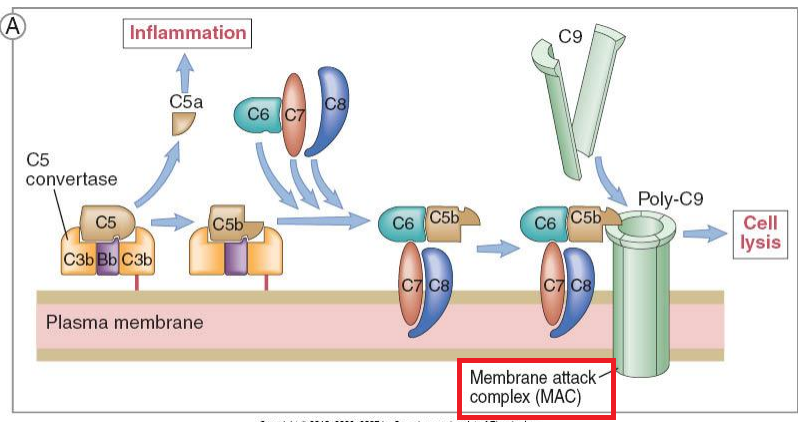
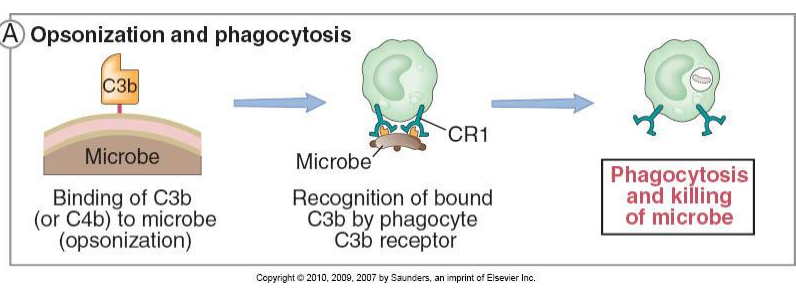
What is the most important biological activity of complement? Define what this activity is and state what complement proteins are involved.
Opsonization: When particulate antigens become coated with C3b, this fragment can interact with specific receptors on phagocytic cells (neutrophils, eosinophils, monocytes, macrophages) triggering adherence and then particle ingestion. This adherence promoting activity is called opsonization. It is the most important biological activity of complement, and is also mediated by breakdown fragments of C3b, called iC3b and C3dg, which bind to different receptors on phagocytes. Clearance of immune complexes is also dependent on C3 fragment deposition.
What enzyme acts on C2b? What is produced? What are the effects?
Inflammation 1 Kinin-like activity: The small fragment released from C2 during activation (C2b) is further degraded by plasmin (a protease in the fibrinolytic system) to release a small (about 20 amino acid) peptide with kinin-like activity. Kinins are vasoactive peptides that cause increased vascular permeability, edema, and pain.
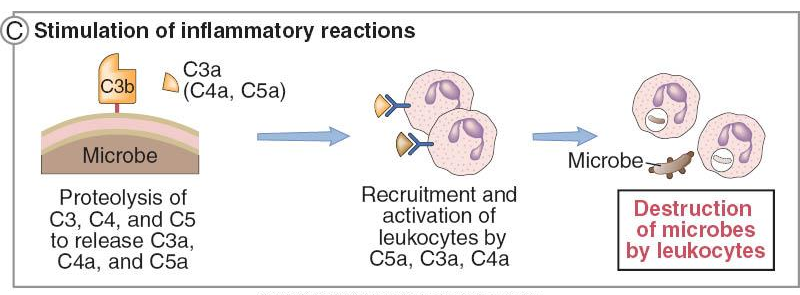
What complement proteins are known as the anaphylatoxins? What are the functions of these proteins?
Inflammation 2 Anaphylatoxins: The small fragments C3a, C4a, and C5a interact with receptors on basophils and tissue mast cells causing granule release. The granules contain a variety of vasoactive and inflammatory mediators including histamine and heparin. The cells also synthesize prostaglandins and leukotrienes which also mediate inflammation. The net effect is smooth muscle contraction, bronchial constriction, and increased vascular permeability.
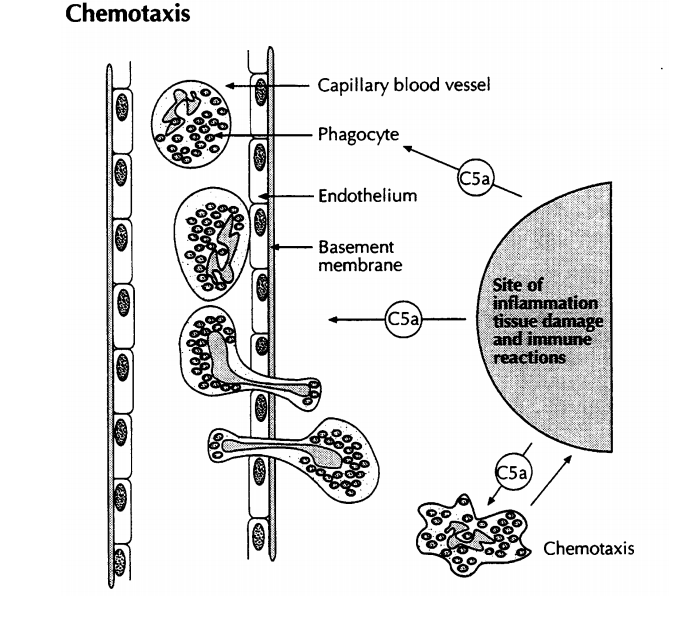
What is the function of C5a? What is its degradation product and what is its function?
Inflammation 3. Chemotaxis: C5a has an additional activity as a chemotactic factor, a chemical that causes directed cell movement. C5a and its degradation product C5a des arg cause neutrophils, eosinophils, monocytes, and macrophages to migrate towards higher concentrations of the molecules, thus accumulating at sites of infection, inflammation, or immune reactions. At high concentrations, C5a also activates the phagocytic cells, making them more metabolically active, and more efficient at destroying the antigen they ingest, and triggering granule enzyme release.
Explain how the alternative pathway is controlled/regulated.

Control of the alternative pathway. Such a potent amplification system obviously needs to be tightly regulated. Without a mechanism to stop the feedback, activation of a small amount of C3 would quickly lead to the consumption of virtually all of the C3 in the body. This is, in fact, just what happens in patients with a genetic deficiency of one of the control proteins.
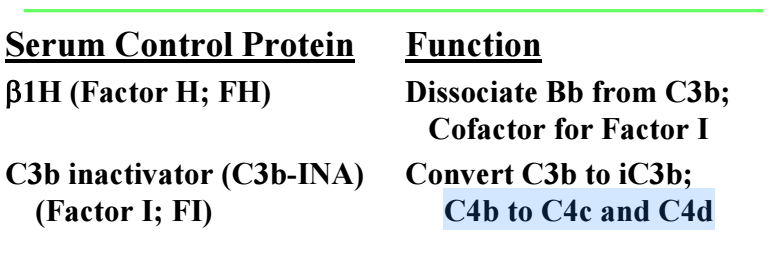
The first control which keeps the alternative pathway in check is the natural lability of the complex enzymes. They decay by loss of Bb, even from the properdin stabilized enzymes. This decay is vastly accelerated by the first control protein, Factor H. Besides this decay accelerating activity, Factor H also works in concert with an enzyme, Factor I (C3b-inactivator), to cleave the C3b into inactive fragments, iC3b, C3c and C3dg. Factor I also converts C4b to C4c and C4d.
C3bBb + H –> C3bH + Bb (inactive)
C3bH ———————> iC3b + H
C3b-INA (I)
What activates the alternative pathway?
The alternative pathway can be activated by many of the complex polysaccharides of yeast or bacterial cell walls, bacterial endotoxins (lipopolysaccharides) and even some viruses and virus infected cells. Certain immunoglobulins also can activate the pathway when they are aggregated. These include IgG1,2,3, IgA, and perhaps IgE. The broad range of activators assures that most extracellular bacterial or fungal pathogens will be able to activate the pathway.
What is the determining factor of alternative pathway activation?
Explain what factor stimulate this pathway to be activated vs inactivated and what controls this?
Mechanisms of activation: The determining factor in alternative pathway activation is the nature of the surface on which C3b is bound. Hydrolysis of C3 is a slow, spontaneous process that steadily generates low levels of C3(H20)Bb in solution. This enzyme is rapidly inactivated by Factors H and I, but while it is active it cleaves a number of C3b molecules which bind covalently, at random, to nearby surfaces. If the surface in a non-activator (normal host cells, some viruses, sheep erythrocytes) Factors I and H rapidly inactivate it, and no biological activities are generated, due to lack of amplification. If the surface is an activator (stated on previous card), structures on the surface protect C3b from the control proteins, leaving it free to bind Factor B and generate the amplification C3 convertase, C3bBbP.
Explain the role of decay in controlling the complement pathways.
Explain the role of how quickly complment proteins bind to their targets (antigens) determines activity and what complement proteins are affected by this.
Decay. The C3 and C5 cleaving enzymes C4b2a, C4b2a3b, C3bBbP, C3(H20)Bb and C(3b)2BbP decay by loss of C2a or Bb by dissociation. They are active for only a short period of time.
The covalent binding of C3b or C4b must occur very quickly, or the thioester bond will react with a water molecule, inactivating the fragment in terms of its ability to bind to an antigen. Similarly, C5b must bind to C6, and the C5b67 complex must bind to a hydrophobic surface within 0.1 sec, or they, too will be inactivated.
What is the function of C1 inhibitor? How does it perform its function?
C1-inhibitor binds C1r and C1s, irreversibly inactivating these enzymes and dissociating C1r and C1s from C1q.
What is the inheritance pattern for complement deficiency? Which complement protein inheritance pattern is the exception?
What alleles have C2 and C4 defienciency been linked to? Factor B?
Family studies have indicated autosomal codominant inheritance of all the C system deficiency genes except properdin, which is X-linked. In addition, genes for C2 and C4 deficiency have been linked to certain HLA alleles, and along with genes for factor B, mapped to a region between HLA-B and HLA-DR on chromosome 6. Their location within the major histocompatibility complex suggests a possible role for C in immunoregulation.
State the consequences of deficiencies to the following proteins:
C1q, C1r, C1s, C2, C3, C4, C5, C6, C7, C8, properdin
(a) deficiency of C1q, C2 or C3 – patients may show increased susceptibility to infection by encapsulated bacteria resulting from decreased serum opsonic activity.
(b) deficiency of C5, C6, C7, C8 or properdin -propensity to Neisserial sepsis (gonococcal, meningococcal), despite normal immunity to other microorganisms, indicating the singular importance of the complement attack complex in protection from Neisseria.
(c) deficiency of a classical pathway component – the incidence of C-mediated dermatitis, arthritis and/or nephritis, including systemic lupus erythematosus and lupuslike diseases, is strikingly elevated in patients lacking an early-acting classical pathway component (C1, C4 or C2; it also is increased, but to a lesser degree, among persons lacking the later-acting components. This may reflect a role for C in clearance of immune complexes or in dealing with certain infectious agents.

What is the consequence of C1-inhibitor defects? How is it treated?
Patients with either deficient secretion of normal C1-INH or with secretion of dysfunctional C1-INH have been described. To date the majority of cases have resulted from genetic abnormalities, with autosomal dominant inheritance, but acquired C1-INH deficiency has been described in a number of patients with lymphoproliferative diseases accompanied by monoclonal gammopathy.
These defects permit uncontrolled, fluid-phase activation of C4 and C2, lowering levels of both and periodically generating sufficient quantities of a kinin-like substance in plasma causing recurrent episodes of angioedema in the skin, viscera and larynx (hereditary angioedema, HAE, or angioedema with acquired C1-INH deficiency).
Treatment with modified androgens (danazol, stanazolol) significantly increases C1-INH and C4 levels and controls attacks. Prior to the introduction of anabolic steroid therapy, treatment with plasmin inhibitors such as EACA was utilized with lesser success, nonetheless emphasizing role(s) of C1-INH as an inhibitor of plasmin and of bradykinin generation via fibrinolysis, as well as of C4 activation. Transfusion replacement therapy utilizing normal plasma has also succeeded, despite theoretical objections to infusing additional C1esterase substrates along with the donor C1-INH.
What are the consequences of deficiencies of Factor I and Factor H?
How is it treated?
Both inborn and acquired deficiencies of C3 regulatory proteins have been described. Patients with selective inborn deficiency of Factor I and Factor H have been described.
In each case hypercatabolism of C3 is the result, and attendant complications are those of C3 deficiency – recurrent pyogenic bacterial sepsis and/or glomerulonephropathy. Treatment is essentially the same as for C3 deficiency: prompt diagnosis and aggressive antibiotic therapy for infections; and for glomerulonephritis, treatment with prednisone or other antiinflammatory agents as indicated.

-
Blood and hematopoiesis week 123
-
Blood, plasma, and serum components week 118
-
Hemostasis week 120
-
Introduction to immunology week 111
-
Innate immunity week 118
-
Cytokines, cytokine receptors, and lipid mediators of immune responses week 121
-
Complement week 121
-
Structure of the immune system: lymphoid tissues and organs week 122
-
Phagocytes and antigen presentation week 113
-
Antibody structure and AbAg interactions week 235
-
T-lymphocyte antigen recognition and MHC week 218
-
Antibody/TCR genetics week 215
-
Lymphocyte differentiation and development week 217
-
Cellular Immunity week 219
-
Antibody Synthesis week 213
-
Memory T and B cells week 212
-
Vaccines week 25
-
Immune regulation and tolerance week 214
-
Mucosal Immunity week 218
-
HIV/AIDS week 217
-
B and T cell immunodeficiency week 327
-
Autoimmunity week 319
-
Allergy week 312
-
Transplantation immunology week 34
