Heritable Bleeding Disorders Flashcards
(79 cards)
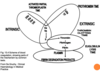
Describe the formation of a haemostatic plug
- Normal platelets in flowing blood
- Damaged endothelium leads to platelets adhering to the wall (bind to collagen in sub endothelium) and becoming activated
- Activated platelets aggregate to form a thrombus (platelet plug)
- Receptors are expressed and fibrinogen binds to platelets
- Granules are released, stimulating further platelet activation
- Coagulation cascade follows – clotting factors are soluble proteins and contribute to fibrin formation.

Which enzyme converts fibrinogen to fibrin?
thrombin (serine protease)
How is thrombin formed?
Prothrombin (coagulation factor II) is cleaved to form thrombin
What is Von Willebrand factor?
Von Willebrand factor helps platelets aggregate and adhere to (collagen in) walls of blood vessels.

What is the most common heritable bleeding disorder?
Von Willebrand disease
What is Von Willebrand disease?
low or dysfunctional Von Willebrand factor –> bleeding
What acts as receptors on platelet surfaces?
Membrane glycoproteins
The membrane glycoproteins of platelets act as receptors that mediate 2 important functions. What are these?
- adhesion to the subendothelial matrix
- platelet aggregation
What are the 2 important membrane glycoproteins found on platelets? What is each one a receptor for?
- 2b3a –> receptor for fibrinogen
- 1b9 –> receptor for vWf
Inside the platelets are granules. What are the 3 different types of granules found?
- alpha granules
- dense granules
- lysosomal granules
What do alpha granules contain?
contain adhesion proteins and calcium (role in coagulation cascade)
What do dense granules contain?
contain ATP and ADP which are released when platelets become activated (ADP forms positive feedback loops – activates more platelets)
Metabolic processes are going on inside cytoplasm of platelets when they become activated. What 2 major metabolic processes are going on?
- Formation of prostaglandins
- Formation of thromboxane
How are prostaglandins formed in the cytoplasm of platelets?
- Mobilisation of fatty acids from platelet membrane
- Conversion of the fatty acid arachidonic acid to intermediate prostaglandins
How is thromboxane formed in the cytoplasm of platelets?
- Synthesised by activated platelets from arachidonic acid (via the enzyme cyclooxygenase)
- Released from phospholipid membrane upon platelet activation
- Secreted into surrounding and stimulates activation of new platelets as well as platelet aggregation
What enzyme converts arachidonic acid to thromboxane?
cyclooxygenase
how does aspirin act as an anti-thrombotic agent?
Aspirin inhibits cyclooxygenase (COX) –> prevents production of thromboxane A2
Name 4 anti-platelet drugs
- Aspirin
- Clopidogrek
- Dipyridamole
- IIb and IIa anatagonists

How does Clopidogrel act as an anti-platelet drug?
- blocks ADP receptor (P2Y12 inhibitor)
- P2Y12 is a chemoreceptor for ADP
How does Dipyridamole act as an anti-platelet drug?
Stimulated prostacyclin to inhibit binding site for collagen, thrombin, thromboxane A2
How do IIb and IIa antagonist act as an anti-platelet drugs?
blocks GPIIb/IIIa
Overview of coagulation cascade
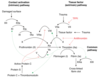
Describe the steps of the extrinsic pathway
- Coagulation is triggered by tissue factor
- Factor VII binds to tissue factor and becomes activated –> VIIa
- VIIa can directly activated X –> Xa

What is tissue factor? Where is it found?
a cellular protein found on surface of cells – damaged endothelium, inflamed blood vessels, WBCs, brain cells