Carnitine Palmitoyltransferase II Deficiency
Type: Disorder of long-chain fatty acid oxidation
Genetics: Autosomal recessive
3 forms: lethal neonatal, infantile hepatocardiomuscular or myopathic form
Clinical Features: Hypoketotic hypoglycemia, liver failure, cardiomyopathy, seizures, peripheral neuropathy
Myopathic: exercise-induced muscle pain and weakness, myoglobinuria
Investigations:
- Molecular gene testing of CPT2
- Reduced CPT enzyme in muscle
- High CK
- High C12-C18 on acylcarnitine profile
Management:
- High carb, low fat diet
- Carnitine
- Glucose infusions during illnesses
- Stay hydrated
- Avoid triggers and VPA, general anesthesia, ibuprofen and diazepam
A child with blonde hair much lighter than her family members is noted to have crystals on her optometry examination.

CYSTINOSIS
Genetics: CTNS gene
Inheritance: Autosomal Recessive
Lysosomal storage disorder leading to cysteine accumulation in all cells
Clinical Features: Normal at birth → failure to thrive, Fanconi syndrome (proximal tubule dysfunction → normal AG metabolic acidosis, volume depletion from polyuria, electrolyte imbalances, hypophosphatemic rickets) by 6-12 months; often fair complexion, photophobia (leads to blindness)
If untreated, ESRD by 10yo.
Investigations:
- Urine: ↑glucose, ↑phosphate, ↑amino acids, ↑organic acids, ↑protein, ↑potassium, ↑bicarbonate
- Serum: ↓phosphate, ↓hypokalemia, non-anion gap metabolic acidosis
- Genetic testing or leukocyte cysteine levels
Management:
- Oral cysteamine - delays onset of complications
- Eyedrops
- Water, sodium, phosphate and potassium supplementation
- Vitamin D
- Bicarbonate supplementation
- Referrals to Nephrology, Metabolics, Ophthalmology, Endocrinology
A hypotonic infant with sweet smelling cerumen
MAPLE SYRUP URINE DISEASE
Presentation: Poor feeding, hyper- or hypotonia, lethargy or encephalopathy
Distinct Features: Maple syrup smelling urine/cerumen
Screening labs: ↑ketones, normal or ↑ ammonia
Acute management:
- IV dextrose
- Branched-chain amino acid-free TPN
- Dialysis PRN
Chronic management:
- Branched-chain amino acid restriction
- Valine and isoleucine supplementation
- Trial of thiamine (rare forms are responsive)
- liver transplantation
A developmentally normal child presents with a hypoglycemic seizure during an episode of gastroenteritis. No ketones are noted on workup.
MCAD DEFICIENCY
Presentation: Identified on Newborn Screen.
Neonatal Form: Hypoglycemia, arrhythmia, cardiac arrest
Management:
- Supplement breast milk with formula and/or expressed breast milk after feeds until supply is clearly established
- With illness: Treat with D10W during acute illness (correct hypoglycemia and suppress lipolysis).
- Avoid fasting (<10-12h)
Tests positive on a Newborn Screen and is associated with ADHD
PHENYLKETONURIA (PKU)
Inheritance: Autosomal Recessive
If untreated, leads to profound intellectual disability.
Maternal PKU is associated with cardiac defects, microcephaly, ID
Clinical Features:
- Hyperactivity with autistic behaviours, including purposeless hand movements, rhythmic rocking and athetosis
- Lighter skin complexion than unaffected siblings
- Seborrheic or eczematoid rash
- Seizures, spasticity, hyperreflexia, tremors (>50% have EEG abnormalities)
- Microcephaly, enamel hypoplasia, growth retardation
- Low bone mineral density, osteopenia
Investigations: once diagnosed, order BH4 studies to rule out deficiency as cause
Management:
- Formula free of phenylalanine
- Ensure tyrosine intake is adequate
- Screen for ADHD - executive dysfunction seen in early-treated children
- Pegvalase (PEG phenylalanine ammonia lyase)
- Liver transplant
Name 5 metabolic disorders that present with an acute presentation?
SMALL MOLECULES
Amino acid disorders: Urea cycle Defects, Maple Syrup Urine Disease
Organic acidemias: Propionic acidemia, MMA
Fatty acid oxidation defects: LCHAD, TFP
Carbohydrate disorders: Galactosemia, GSD
Energy Defects: Lactic acidosis
Vitamins: Biotinidase, pyridoxine dependent seizures
Metals: Menke’s
Name 5 metabolic disorders that present with a chronic presentation?
ORGANELLES (MACROMOLECULES)
Lysosomes:
- CNS: Tay Sachs, Krabbe, Batten’s
- Coarse facial features: MPS, Oligosaccharides (Hurler, Sialidosis)
- Organs: Gaucher, Niemann Pick
Peroxisomes: Zellweger, Adrenoleukodystrophy
Mitochondria: MELAS, MERRF, NARP, LHON
Golgi/ER: Congenital disorders of glycosylation
Membranes: Transporters/Trafficking
Macromolecule synthesis/degradation: Lipidomics, protein translation
List 5 components of an initial metabolic work-up.
Blood:
Blood gas
Electrolytes (with anion gap calculation)
Glucose
Ammonia
Lactic acid
Plasma/Serum Amino Acids
Acylcarnitine
Total/free carnitine
Urine:
Urinalysis - for ketones
Urine Organic Acids
List 2 causes of Nonketotic Hypoglycemia
What is the best test to define the diagnosis?
What is the immediate treatment?
↓insulin, ↑FFA:
- Fatty acid oxidation defect, ketogenic defect
↑insulin, ↓FFA:
- Hyperinsulinism
Plasma Acylcarnitine
Immediate treatment: glucose infusion
List 2 causes of Ketotic Hypoglycemia
What is the immediate treatment?
NO Hepatomegaly:
Organic aciduria, Ketolytic defect
YES Hepatomegaly:
Glycogen storage disease, Gluconeogenesis defect
Immediate treatment: glucose infusion
Name 2 causes of hyperammonemia
Describe the immediate treatment.
- Organic aciduria (metabolic acidosis, big AG)
- Fatty acid oxidation defect (hyperchloremic acidosis, little/no AG +/- hypoglycemia)
- Urea Cycle Defect (respiratory alkalosis, little/no AG)
- Citrullinemia
- Arginase deficiency
- OTC deficiency
Treatment
- Protein restriction
- Ammonia removal (sodium benzoate and sodium phenylbutyrate, dialysis)
- Arginine to drive urea cycle (except in arginase deficiency)
Name 1 metabolic disorder that causes an elevated lactate level.
Organic acidopathies
Fatty acid oxidation defects (acylcarnitine profile)
Glycogen storage disease (GSD I)
What metabolic disorder tends to present with the following lab abnormalities in a 4 day old male?
ABG: 7.5/25/20
Na 140, Cl 104, Glucose 4, AG 16
What additional investigation would help narrow your differential?
What additional investigation would confirm your diagnosis?
Urea Cycle Disorder
Ammonia level (ammonia 800µmol/L)
Plasma Amino Acids
List 3 options for the management of a Urea Cycle Disorder
Protein restriction
IV glucose infusion (stop catabolism)
Replace arginine (may be playing a role in encephalopathy)
Remove ammonia (sodium benzoate, sodium phenylacetate)
Dialysis - if required
What metabolic disorder tends to present with the following lab abnormalities in a 4 day old male?
ABG: 7.0/25/5
Na 140, Cl 103, Glucose 4, AG 32, ammonia 800µmol/L
What test would confirm your diagnosis?
Organic acidemia (metabolic acidosis with big AG)
Urine Organic Acids
What metabolic disorder tends to present with the following lab abnormalities in a 4 day old male?
ABG: 7.2/25/14
Na 140, Cl 114, Glucose 2, AG 12, ammonia 800µmol/L
What test would confirm your diagnosis?
Fatty Acid Oxidation Defect (hyperchloremic acidosis, little/no AG, hypoglycemia)
Acylcarnitines
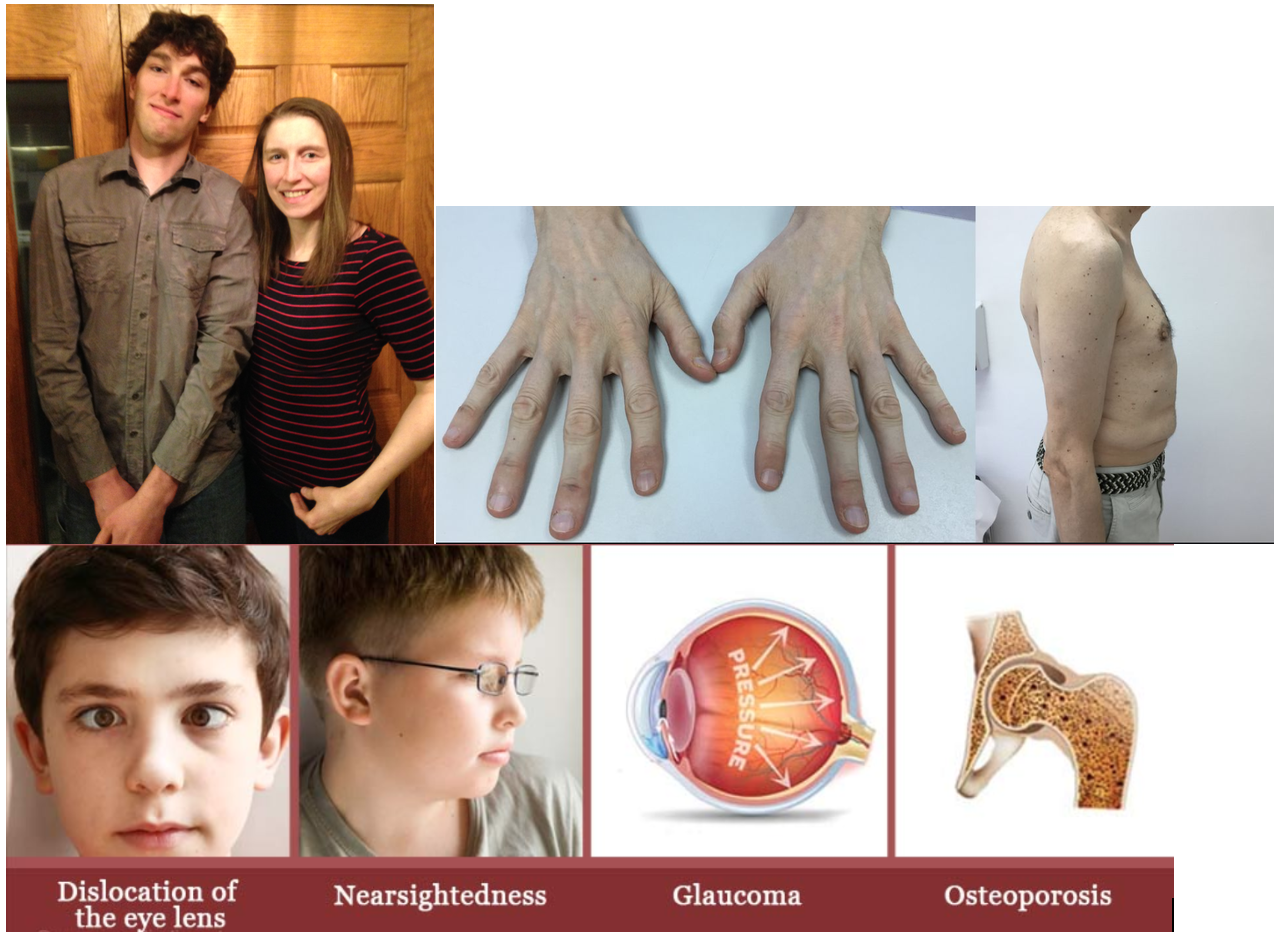
HOMOCYSTINURIA due to CβS-deficient Homocystinuria
Clinical Features: ectopia lentis, myopia, osteoporosis, kyphoscoliosis, skeletal dysplasia, high palate, long fingers (Marfanoid), developmental delay, psychiatric issues, thrombosis (arterial and venous), fair, thin skin, myopathy, peripheral neuropathy
Management:
- Pyridoxine (250-500mgs/day)
- Protein restriction
- Betaine
- ASA, Heparin subcutaneously
- Pregnancy management
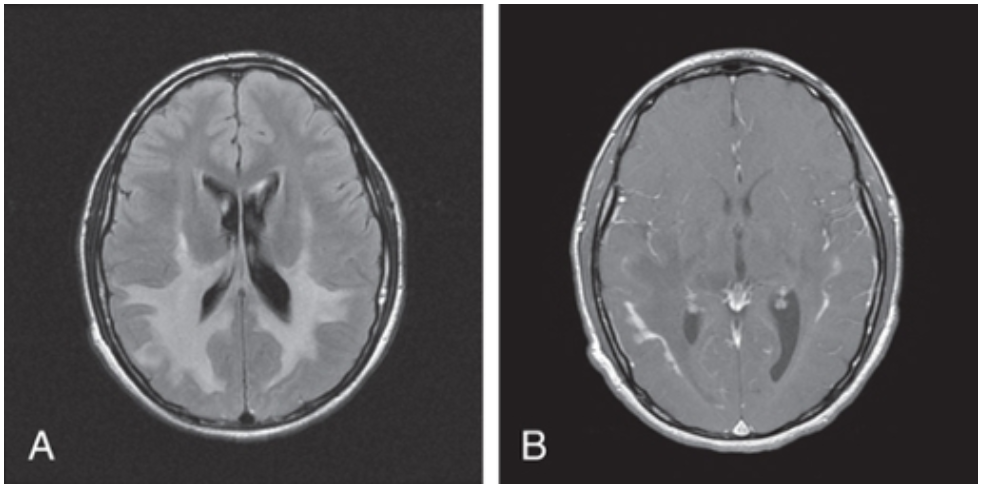
X-LINKED ADRENOLEUKODYSTROPHY
Metabolic disorder: Peroxisomal
Genetics: ABCD1 gene
Inheritance: X-linked recessive
Clinical Features: Onset 4-12yo, developmental regression (classically starting with handwriting, accompanied by behavioural deterioration [hyperactivity, ADHD]), neurologic deterioration with ataxia, seizures and paraplegia, adrenal insufficiency, impaired auditory discrimination
Investigations: Elevated VLCFAs, MRI Brain (occipital demyelination with leading edge enhancement after gadolinium administration), molecular genetic testing
Management:
- Adrenal steroid hormone supplementation
- Bone marrow transplantation
- Lorenzo’s Oil - previously a treatment - has fallen out of favour
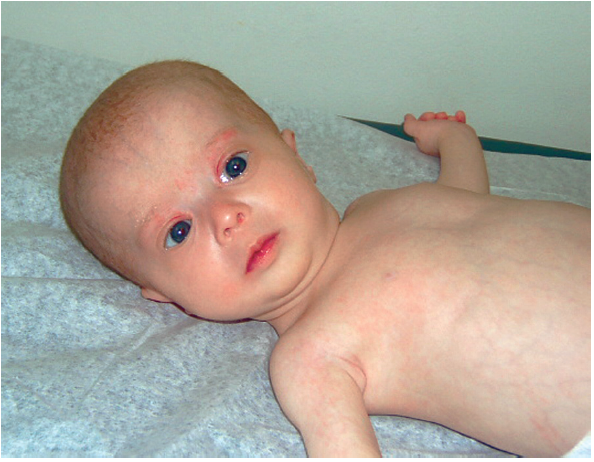
ZELLWEGER SYNDROME
Metabolic disorder: Peroxisomal
Genetics: several genes involved
Inheritance: Autosomal Recessive
Clinical Features: Facial features (high forehead, upslanting palpebral fissures, hypoplastic supraorbital ridges, epicanthal folds), severe weakness and hypotonia, neonatal seizures, eye abnormalities, jaundice
Investigations: Elevated VLCFAs = screening test; AUS for hepatomegaly, renal cysts; molecular genetic testing for known genes involved
Management: Supportive
A metabolic disorder associated with stroke-like episodes before 40-years-old.
MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes)
Metabolic disorder: Mitochondrial
Genetics: several involved (including MT-TL1)
Inheritance: Mitochondrial
Clinical Features: normal initial development, symptoms start in childhood with stroke-like episodes, muscle weakness, headaches, vision impairment, vomiting, WPW, conduction abnormalities, LVH, single endocrine disorders (T1DM
What metabolic disorder is associated with hypertrophic cardiomyopathy and peripheral neuropathy?
LEIGH SYNDROME
Metabolic disorder: Mitochondrial
Genetics: several involved (including MT-TL1)
Inheritance: Autosomal Recessive
Clinical Features: lactic acidosis, FTT, hypotonia, peripheral neuropathy and hypertrophic cardiomyopathy , seizures
Investigations: mtDNA molecular genetic testing, ↑blood/↑CSF lactate levels, gas - metabolic acidosis, muscle biopsy, respriatory chain enzyme studies; MRI brain - bilateral symmetric hypodensitites in the basal ganglia
Management:
- Suppportive management
- Routine surveillance of new symptoms
- Sodium bicarbonate to treat acidosis
- Antiepileptic drugs to treat seizures (avoid VPA)
- Multidisciplinary team management is encouraged
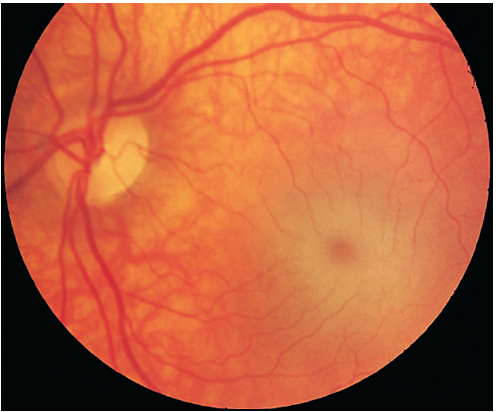
GM2 GANGLIOSIDOSIS - TAY-SACHS DISEASE
Metabolic disorder: Lysosomal lipid storage disorder
Genetics: HEXA gene
Inheritance: Autosomal Recessive
Most prevalent in Ashkenazi Jewish population
Clinical Features: Hyperacusis, no HSM.
- Onset 4-6m. Previously normal development with marked startle reaction, then developmental delay followed by regression.
- By 1yo, lose ability to stand, sit and vocalize + macrocephaly becomes apparent (d/t ↑GM2 ganglioside deposition in brain)
- Hypotonia → spasticity → convulsions, blindness, deafness and bilateral cherry-red spots
Investigations: serum or leukocyte β-hexosaminidase A deficiency
Prognosis: few live beyond 3-4yo; death usually due to aspiration or pneumonia
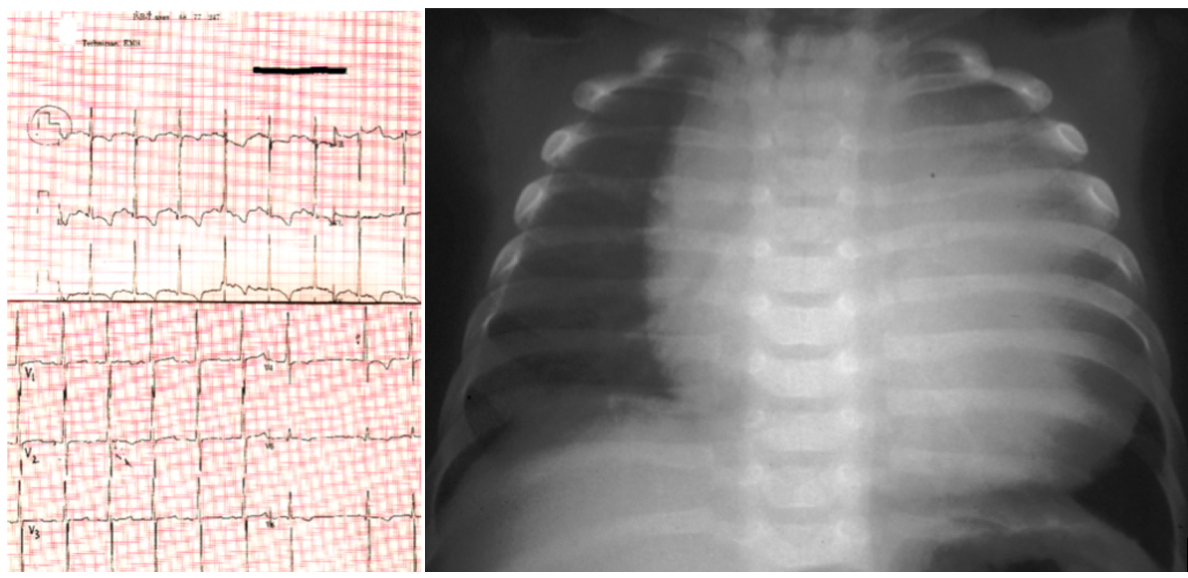
POMPE DISEASE
Metabolic disorder: Glycogen Storage Disease
Genetics: GAA gene
Inheritance: Autosomal Recessive
Clinical Features: Hypertrophic cardiomyopathy, FTT, severe hypotonia, short PR interval, macroglossia
Investigations: muscle biopsy (glycogen-containing vacuoles), ECG (shortened PR, large QRS), enzyme studies, gene studies, urine hex4 (tetrasaccharide) - level correlates with severity
Management: Enzyme replacement therapy
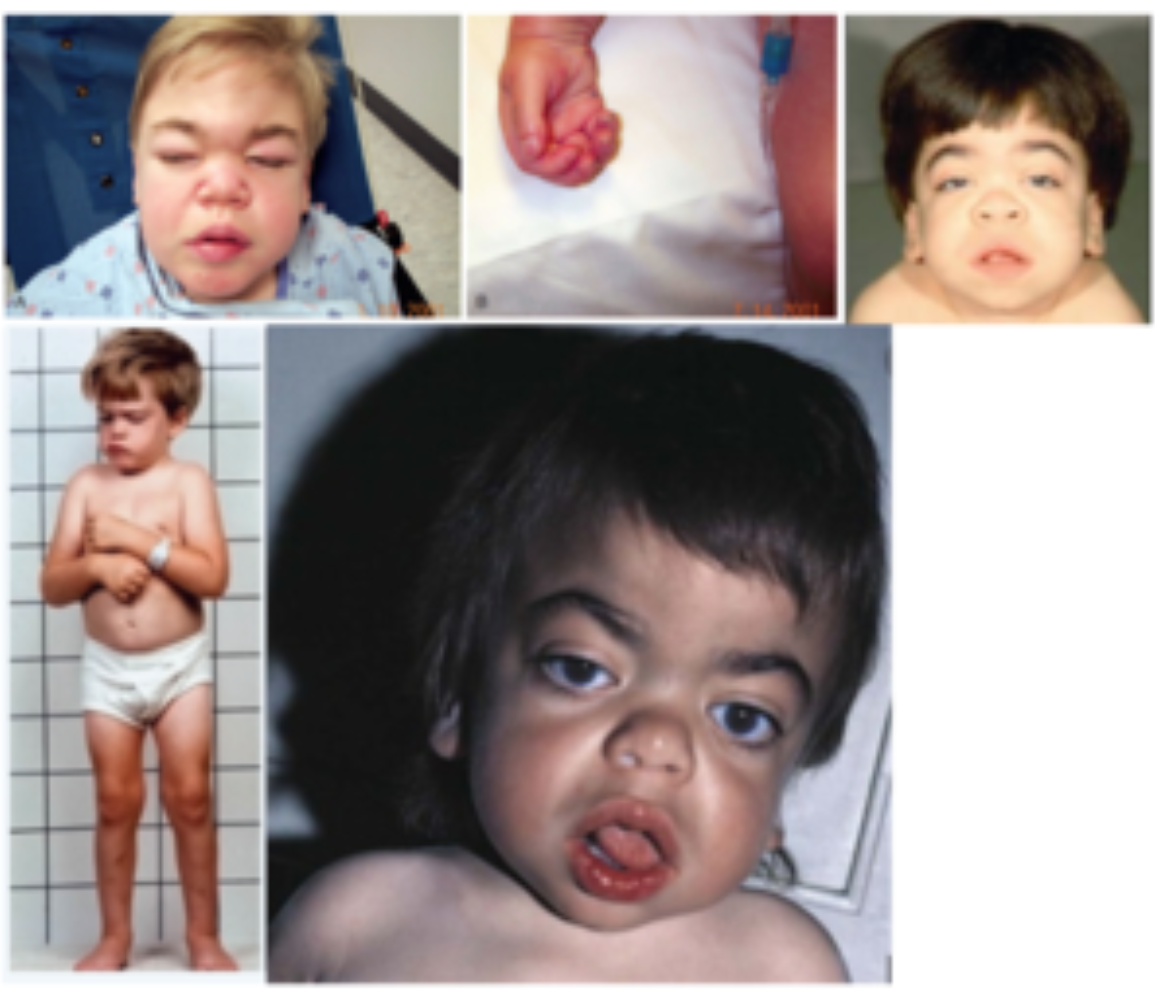
HURLER SYNDROME (MUCOPOLYSACCHARIDOSES [MPS1])
Metabolic disorder: Lysosomal Disorder
Genetics: IDUA 4p16.3
Inheritance: Autosomal recessive
MPSI is a spectrum from Hurler (severe) to Scheie syndromes (least severe)
If untreated, Hurler is fatal in 5-10y
Clinical features: ID, coarse facial features, corneal clouding, visceromegaly, short stature, joint contractures, dysostosis multiplex, leucocyte inclusions, mucopolysacchariduria
Investigations: Gene testing, enzyme assays, urine glucosaminoglycans
Management:
- BMT can help disease (if done before CNS symptoms) - usually <1yo
- Enzyme supplementation for non-CNS disease (skeletal penetrance is poor)

-
Acute Care270
-
Adolescent/Gyne98
-
Allergy/Immunology70
-
Cardiology136
-
CYPT and Ethics64
-
Dermatology70
-
Endocrinology161
-
ENT/Ophtho/Gen Surg149
-
Gen Paeds0
-
Genetics179
-
GI/Nutrition146
-
Heme/Onc106
-
Infectious Disease271
-
Metabolics27
-
MSK/Ortho/Rheumatology138
-
Nephrology/Urology154
-
Neurology110
-
NICU233
-
Pharm and Critical Appraisal57
-
Psych/Development200
-
Respirology152
-
ADHD13
-
Syndromes102
