UW7 Flashcards
SLE Antibodies
Antinuclear Antibodies (ANA) sensitive
Anti-DSDNA antibodies & Anti-Smith antibodies (specific)
Lupus nephritis is caused by
type III hypersensitivity reaction, immune-complexes deposition in the mesangium
Ankylosing spondylitis description
Ankylosing spondylitis is a chronic inflammatory arthritis involving the spine and sacroiliac joints. It presents with low back pain and stiffness. Common associated features include enthesitis, dactylitis, and uveitis
Question

This patient is pregnant with twins of different sexes which can only occur in dizygotic twins due to the fertilization of 2 oocytes by 2 different sperm. Dizygotic twins are almost always dichorionic/diamniotic (eg, 2 chorions and 2 amnions) but may falsely give the appearance of being monochorionic/monoamniotic if the chorions and amnions fuse due to proximity of implantation sites.

Dizygotic twins
Dizygotic twins are almost always dichorionic/diamniotic (eg, 2 chorions and 2 amnions) but may falsely give the appearance of being monochorionic/monoamniotic if the chorions and amnions fuse due to proximity of implantation sites

Monozygotic twins
monozygotic twins develop from division of a single zygote after fertilization of a single oocyte. They are of the same sex, are genetically identical, and are similar in appearance. The type of placentation in monozygotic twins depends on when zygote division occurs during embryonic development.
Early division (days 0-4) can result in monozygotic twins with 2 chorions and 2 amnions (eg, dichorionic/diamniotic), which may or may not be fused. If the sexes are the same, it may be difficult to distinguish whether the twins are monozygotic or dizygotic until later in the pregnancy.
Division between days 4-8 is the most common outcome in monozygotic twins and results in 1 chorion (eg, shared placenta) but 2 amnions (eg, monochorionic/diamniotic twins)
Late division (8-12 days) results in 1 chorion and 1 amnion. A monochorionic/monoamniotic pregnancy is associated with a high fetal fatality rate, due primarily to the increased risk of umbilical cord entanglement
Division occurring after 13 days can result in monochorionic/monoamniotic conjoined twins

Monozygotic twins: Early division (days 0-4)
Early division (days 0-4) can result in monozygotic twins with 2 chorions and 2 amnions (eg, dichorionic/diamniotic), which may or may not be fused. If the sexes are the same, it may be difficult to distinguish whether the twins are monozygotic or dizygotic until later in the pregnancy.

Monozygotic twins: Division between days 4-8
Division between days 4-8 is the most common outcome in monozygotic twins and results in 1 chorion (eg, shared placenta) but 2 amnions (eg, monochorionic/diamniotic twins)

Monozygotic twins: Late division (days 8-12)
Late division (8-12 days) results in 1 chorion and 1 amnion. A monochorionic/monoamniotic pregnancy is associated with a high fetal fatality rate, due primarily to the increased risk of umbilical cord entanglement

Monozygotic twins: Division > or = 13 days
Division occurring after 13 days can result in monochorionic/monoamniotic conjoined twins

Question

C. polymorphic ventricular tachycardia (torsade du pointes)
Relatively young patients with symptomatic paroxysmal atrial fibrillation are often treated with a rhythm-control strategy to restore and maintain normal sinus rhythm and to eliminate symptoms. Antiarrhythmic drugs commonly used for rhythm control of atrial fibrillation include class IC (eg, flecainide, propafenone) and class III (eg, ibutilide, dofetilide) agents.
Dofetilide and ibutilide selectively block the rapid component of the delayed-rectifier potassium current. This slows repolarization and prolongs action potential duration and the effective refractory period in cardiomyocytes, suppressing the electrical foci that lead to atrial fibrillation. As a result, the QT interval is prolonged, leading to an increased risk of torsade de pointes, a form of polymorphic ventricular tachycardia that can cause syncope and sudden cardiac death. To minimize this risk, dofetilide or ibutilide therapy is started only in the hospital with temporary cardiac monitoring.
(Choice A) Adenosine can trigger bronchospasm, likely by stimulating the release of histamine and leukotrienes from mast cells. Class III antiarrhythmics are not associated with bronchospasm.
(Choice B) Beta blockers (via inhibition of beta-1 receptor–driven chronotropy) and nondihydropyridine calcium channel blockers (via blockade of L-type atrioventricular [AV] node calcium channels) slow AV node conduction and in overdose can cause complete AV block. Digoxin (via enhanced vagal tone) and adenosine can also cause AV block. Although some class III antiarrhythmics (ie, sotalol, amiodarone) have negative chronotropic effects and may slow AV node conduction, dofetilide and ibutilide have no significant effect on AV node conduction.
(Choice D) Amiodarone can disrupt thyroid function and lead to hyper- or hypothyroidism, but dofetilide and other class III antiarrhythmics do not affect thyroid function. Compared with other class III antiarrhythmics, amiodarone is associated with a lower risk of torsade de pointes.
(Choice E) Most antiarrhythmic drugs, including class III, are not associated with an increased risk of venous thromboembolism.
(Choice F) Digoxin toxicity can cause visual disturbances consisting of blurred or yellow vision, but visual disturbance is not an adverse effect of class III antiarrhythmics.
Educational objective: Dofetilide and ibutilide are class III antiarrhythmics that treat atrial fibrillation by blocking the rapid component of the delayed-rectifier potassium current to slow repolarization and increase the effective refractory period. As a result, the QT interval is prolonged, creating an increased risk of polymorphic ventricular tachycardia (torsade de pointes).
Question

E.

How to organize a table into standard format

Question

B. Effect modification
Effect modification occurs when the effect of an exposure on an outcome is modified by another variable. It can be identified using stratified analysis (analyzing the cohort as different subgroups), as the different strata will have different measures of association. In this scenario, smoking status modified the effect of the new estrogen receptor agonist (exposure) on deep vein thrombosis (DVT) incidence (outcome). Using stratified analysis by smoking status:
Among smokers, there was a statistically significant association between taking the new estrogen receptor agonist and risk of developing DVT with a relative risk of >1, indicating higher risk, and a p-value of <0.05, indicating statistical significance.
In contrast, among nonsmokers, there was no statistically significant association between taking the medication and risk of DVT (p-value >0.05).
Effect modification is not a bias (Choices D and E), as it is not due to flaws in the design or analysis phases of the study. It is a natural phenomenon that should be described, not corrected.
Effect modification is most easily confused with confounding (Choice A), but stratified analysis can help distinguish between these 2 scenarios. With effect modification, the different strata will have different measures of association, as seen in this example of the association between taking the estrogen receptor and the risk of DVT among smokers compared to nonsmokers. In contrast, with confounding, stratification usually reveals no significant difference between the strata. For instance, in an analysis of primary school students (of all grade levels), age can be a confounder that muddies the association between shoe size and intelligence. Children with bigger shoe sizes may appear to be more intelligent on initial analysis. However, this association is likely not due to shoe size but rather to age because older children tend to have both bigger feet and more intelligence. When older and younger children are analyzed separately (stratification based on the confounder), the association between shoe size and intelligence disappears.
(Choice C) The latent period is the time required for an exposure to begin having an effect. However, there is no information on how latency was handled in this study.
Educational objective:
Effect modification is present when the effect of the main exposure on the outcome is modified by the presence of another variable. Effect modification is not a bias.
Question

C. Neisseria Meningitidis

Paroxysmal nocturnal hemoglobinuria (PNH) is caused by a mutation in hematologic stem cells that eliminates production of an anchoring protein that attaches surface molecules to the cell membrane. Loss of this membrane anchor prevents erythrocytes from expressing the complement-inactivating surface proteins CD55 and CD59. Without these surface proteins, autoactivated components of the alternative complement cascade cannot be disabled. This leads to spontaneous complement amplification, the generation of membrane attack complexes (MACs) on the red cell membrane, and subsequent complement-mediated hemolysis.
Treatment with a monoclonal antibody (eg, eculizumab) that targets C5, the first complement protein that contributes to the formation of MACs, can drastically reduce hemolysis and improve symptoms (eg, fatigue) in patients with PNH. However, the generation of MACs is crucial for defense against encapsulated organisms (eg, Neisseria meningitidis, Streptococcus pneumoniae) because these pathogens are resistant to other host defense mechanisms (eg, immunoglobulin binding, phagocytosis).
Patients receiving anti-C5 therapy require vaccination against encapsulated pathogens (N meningitidis, S pneumoniae) and appropriate antibiotic prophylaxis (eg, penicillin) to prevent fulminant infection.
(Choice A) Nontypable strains of Haemophilus influenzae are not encapsulated; therefore, these strains are susceptible to phagocytosis and immunoglobulin-mediated opsonization. Control is not primarily mediated by the formation of MACs.
(Choice B) Host defense to Mycobacterium tuberculosis depends more on T-cell and antigen-presenting cells than on complement activation and MAC formation.
(Choices D and E) Staphylococcus aureus and Streptococcus pyogenes often secrete factors that disable the complement cascade. Therefore, they are primarily countered by other components of the immune system (eg, neutrophils, humoral immunity).
Educational objective:
Paroxysmal nocturnal hemoglobinuria leads to the formation of membrane attack complexes on erythrocytes. Treatment with monoclonal antibody against C5, the first component of the membrane attack complex, can improve symptoms. However, it also increases risk for encapsulated bacterial infection. Therefore, patients require vaccination and antibiotic prophylaxis against Neisseria meningitidis and Streptococcus pneumoniae.
Paroxysmal nocturnal hemoglobinuria Pathophysiology
Paroxysmal nocturnal hemoglobinuria (PNH) is caused by a mutation in hematologic stem cells that eliminates production of an anchoring protein that attaches surface molecules to the cell membrane. Loss of this membrane anchor prevents erythrocytes from expressing the complement-inactivating surface proteins CD55 and CD59. Without these surface proteins, autoactivated components of the alternative complement cascade cannot be disabled. This leads to spontaneous complement amplification, the generation of membrane attack complexes (MACs) on the red cell membrane, and subsequent complement-mediated hemolysis.
Paroxysmal nocturnal hemoglobinuria Tx
Treatment with a monoclonal antibody (eg, eculizumab) that targets C5, the first complement protein that contributes to the formation of MACs, can drastically reduce hemolysis and improve symptoms (eg, fatigue) in patients with PNH. However, the generation of MACs is crucial for defense against encapsulated organisms (eg, Neisseria meningitidis, Streptococcus pneumoniae) because these pathogens are resistant to other host defense mechanisms (eg, immunoglobulin binding, phagocytosis).
Patients receiving anti-C5 therapy require vaccination against encapsulated pathogens (N meningitidis, S pneumoniae, typeable H. flu) and appropriate antibiotic prophylaxis (eg, penicillin) to prevent fulminant infection.
Question

Vitamin D is derived from food and generated in the skin due to sunlight exposure (ie, conversion of 7-dehydrocholesterol to cholecalciferol). Endogenous production from sunlight accounts for approximately half of daily requirements (~200-400 IU/day [required: 600]). The dietary share is usually obtained primarily from fortified dairy/dairy-alternative foods; oily fish (eg, salmon, herring) is also an abundant source. Other foods contain vitamin D but are often insufficient to meet requirements.
Vitamin D deficiency reduces intestinal absorption of calcium. Parathyroid hormone secretion increases to mobilize calcium from bone and maintain plasma calcium levels but causes renal phosphate wasting. The resulting hypophosphatemia causes impaired mineralization of bone. In children, this manifests as rickets, characterized by excessive unmineralized osteoid matrix at the epiphyseal (growth plate) cartilage.
Clinical manifestations of rickets include frontal bossing, craniotabes (softened skull bones), and costochondral widening from cartilage overgrowth (rachitic rosary). Dental enamel hypoplasia can also be seen. Weight-bearing children may show lower extremity bowing (genu varum). Radiographs of growth plates (eg, distal ulna) often reveal metaphyseal plate widening and cupping.
(Choice A) Glucocorticoids (eg, prednisone) cause osteoporosis because they inhibit proliferation and differentiation of osteoblast precursors and increase apoptosis of mature osteoblasts. Fragility fractures may occur, but this patient’s deformities are more consistent with rickets.
(Choice B) The early osteolytic phase of Paget disease of bone is characterized by proliferation of abnormally large, hypernucleated osteoclasts. Paget disease causes bone pain and deformity (eg, frontal bossing, bowing of long bones) but findings are typically focal and asymmetric and usually occur in adults age >50.
(Choice C) Replacement of bone with disorganized fibrous connective tissue (fibrous dysplasia) can occur in a single bone (eg, femur) or multiple bones (polyostotic). Polyostotic fibrous dysplasia (McCune-Albright syndrome) is typically associated with café au lait macules and pituitary endocrine disorders (eg, precocious puberty, thyrotoxicosis, acromegaly).
(Choice D) Osteosarcoma, the most common malignant bone tumor in children, is characterized by excessive production of mineralized bone. The lesions are typically single and would not cause short stature, frontal bossing, or enamel defects.
Educational objective:
Vitamin D deficiency decreases intestinal absorption of calcium. Parathyroid hormone secretion increases to maintain plasma calcium, which causes renal phosphate wasting and impaired bone mineralization. In children, this causes rickets characterized by excessive unmineralized osteoid matrix at the epiphyseal cartilage.

Physical manifestations of Ricketts

Question

E. interchain cross-links involving lysine
This patient’s emphysema is likely due to alpha-1 antitrypsin deficiency. Neutrophil-secreted elastase is an endogenous proteolytic enzyme that hydrolyzes elastin within alveolar walls. The liver synthesizes alpha-1 antitrypsin, a protein that inhibits neutrophil elastase and prevents alveolar wall degradation, particularly in the lower airways. Patients with alpha-1 antitrypsin deficiency consequently develop excessive alveolar elastin degradation, which clinically manifests with early-onset, lower lobe–predominant emphysema.
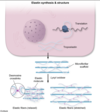
Elastin is a fibrous connective tissue protein that provides elasticity to the skin, blood vessels, and pulmonary alveoli. The fibers can stretch to several times their length and recoil back to their original size once stretching forces are withdrawn. Elastin assembly is closely related to that of collagen. Similar to collagen, elastin is synthesized as a large polypeptide precursor (tropoelastin) composed of about 700, mostly nonpolar, amino acids (eg, glycine, alanine, valine) (Choice D). Elastin also contains proline and lysine residues; however, in contrast to those found in collagen, few of these amino acids are hydroxylated (Choice C).
After tropoelastin is formed, it is secreted into the extracellular space where it interacts with microfibrils (fibrillin) that function as a scaffold. Next, lysyl oxidase, a copper-dependent enzyme, oxidatively deaminates some of the lysine residues of tropoelastin, facilitating the formation of desmosine cross-links between neighboring polypeptides that hold the elastin molecules together. These cross-links, along with the high content of nonpolar (hydrophobic) amino acids, account for the rubber-like properties of elastin.
(Choices A and B) Disulfide bridges are formed during collagen, not elastin, synthesis. After post-translational hydroxylation and glycosylation of procollagen molecules, disulfide bond formation between the C-terminal propeptide regions of 3 alpha chains brings the chains into a favorable alignment for triple helix assembly.
Educational objective:
The rubber-like properties of elastin are due to high content of nonpolar (hydrophobic) amino acids and extensive cross-linking between elastin monomers facilitated by lysyl oxidase. Patients with alpha-1 antitrypsin deficiency can develop early-onset, lower lobe–predominant emphysema due to excessive alveolar elastin degradation.
Question

D. Tyrosine to fumarate

Alkaptonuria is an autosomal recessive disorder of tyrosine metabolism. Deficiency of homogentisic acid dioxygenase blocks homogentisic acid metabolism, preventing the conversion of tyrosine to fumarate. Homogentisic acid accumulates in the body and is excreted in the urine, imparting a black color to the urine if allowed to sit and undergo oxidation. In patients with alkaptonuria, the retained homogentisic acid selectively binds to collagen in connective tissues, tendons, and cartilage. This leads to “ochronosis,” a blue-black pigmentation most evident in the ears, nose, and cheeks, and ochronotic arthropathy, which typically manifests during adulthood.
(Choice A) Leucine is a branched-chain amino acid that is elevated in maple syrup urine disease. Isoleucine and valine are also increased. Impaired metabolism of these amino acids leads to cerebral edema, seizures, and a sweet smell of the urine.
(Choice B) Conversion of phenylalanine to tyrosine is defective in phenylketonuria and usually occurs due to a defect in phenylalanine hydroxylase. Undiagnosed and untreated phenylketonuria results in significant intellectual disability not seen in this patient.
(Choice C) Impaired renal cystine (a homodimer of cysteine) transport leads to cystinuria, a disease characterized by flank pain, hematuria, and renal stones in childhood or adolescence.
(Choice E) Sickle cell anemia results from the substitution of valine for glutamic acid due to a single-nucleotide polymorphism. This mutation leads to loss of red cell elasticity, polymerization of sickle hemoglobin, and sickling of red blood cells, which results in vasoocclusive crises.
Educational objective:
Alkaptonuria is an autosomal recessive disorder in which the lack of homogentisic acid dioxygenase blocks the metabolism of tyrosine, leading to an accumulation of homogentisic acid. Clinical features include a black urine color when exposed to air, a blue-black pigmentation on the face, and ochronotic arthropathy.
Alkaptonuria pathophysiology
autosomal recessive disorder of tyrosine metabolism.
Deficiency of homogentisic acid dioxygenase blocks homogentisic acid metabolism, preventing the conversion of tyrosine to fumarate.
Homogentisic acid accumulates in the body and is excreted in the urine, imparting a black color to the urine if allowed to sit and undergo oxidation.
In patients with alkaptonuria, the retained homogentisic acid selectively binds to collagen in connective tissues, tendons, and cartilage. This leads to “ochronosis,” a blue-black pigmentation most evident in the ears, nose, and cheeks, and ochronotic arthropathy, which typically manifests during adulthood.

Question

A. Expanded tri-nucleotide repeats

This child with intellectual disability has weakness and slow muscle relaxation, findings suggestive of myotonic dystrophy (DM). DM is an autosomal dominant muscular dystrophy due to an expansion of CTG trinucleotide repeats within the dystrophia myotonica protein kinase (DMPK) gene. Increased repeat length occurs with successive generations and is associated with earlier onset of more severe disease (ie, anticipation); this patient’s parent with mild muscle weakness is most likely due to adult-onset DM.
In the childhood form of DM, intellectual disability and behavioral problems are often the predominant features. Other key findings characteristic of classic DM may be present or develop over time:
Weakness, atrophy, and pain of facial and distal limb musculature
Myotonia, or delay in muscle relaxation, such as sustained hand contraction after attempted grip release or sustained thumb abduction after thenar eminence percussion (as seen here)
Cardiac abnormalities (eg, conduction disturbances, cardiomyopathy)
Gastrointestinal tract disturbances (eg, dysphagia, constipation)
Nonmuscular involvement such as frontal hair loss, cataracts (lens opacities), insulin resistance, and daytime somnolence
(Choice B) Mitochondrial myopathies are often maternally inherited and characterized by muscle weakness (eg, exercise intolerance) and lactic acidosis in more severe cases. Cataracts may be present, but myotonia does not occur.
(Choice C) Partial chromosome deletion can be seen with inherited causes of intellectual disability (eg, cri-du-chat syndrome) but would not explain this patient’s myotonia or cataracts.
(Choice D) Uniparental disomy is the mode of inheritance for Prader-Willi and Angelman syndromes, which are associated with cognitive dysfunction and behavioral disturbances but not myotonia.
(Choice E) X-linked frameshift mutation in dystrophin gene can cause both Duchenne and Becker muscular dystrophy. These conditions present with muscle weakness, but the proximal lower extremities are typically affected first and cataracts and myotonia are not seen.
Educational objective:
Myotonic dystrophy is an autosomal dominant condition caused by a trinucleotide repeat expansion; successive generations typically have an increased number of repeats, resulting in earlier and more severe disease (ie, anticipation). In children, cognitive/behavioral issues may be the initial findings before development of muscle weakness and myotonia.
Clinical presentation of Myotonic Dystrophy in childhood
In the childhood form of myotinc dystrophy (DM), intellectual disability and behavioral problems are often the predominant features. Other key findings characteristic of classic DM may be present or develop over time:
Weakness, atrophy, and pain of facial and distal limb musculature
Myotonia, or delay in muscle relaxation, such as sustained hand contraction after attempted grip release or sustained thumb abduction after thenar eminence percussion (as seen here)
Cardiac abnormalities (eg, conduction disturbances, cardiomyopathy)
Gastrointestinal tract disturbances (eg, dysphagia, constipation)
Nonmuscular involvement such as frontal hair loss, cataracts (lens opacities), insulin resistance, and daytime somnolence