ML Flashcards
(230 cards)
ML: What are loss functions?
Functions that numerically compare your predictions Y* to the true values Y to provide feedback on the accuracy of your trained model.
ML: What is the typical loss function for classification? For regression?
Classification: 0/1 loss
Regression: Mean-Squared Error (MSE)
ML: What is a generative model?
It models P(Y|X) using P(X|Y)P(Y)
(derived from Bayes rule)
ML: What is a discriminative model?
Models P(Y|X) directly, as P(Y|X)
ML: What are some advantages of discriminative model over generative? What about generative over discriminative?
Discriminative models (Y|X) don’t need to make assumptions about the distribution of X|Y, and can be simpler as a result.
Generative models can be used to generate samples. They are also sometimes more intuitive.
ML: What is a Bayes classifier?
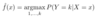
ML: What are 2 important characteristics of a Bayes classifier?
It is the best possible classifier
Its error is thus the irreducible error of a classification problem.
ML: At a high level, how would a Bayes Classifier be constructed in a case where errors are asymmetric, meaning some errors are worse than others?
We would weight each possible error with its own amount of loss Li,j, pertaining to what you said the answer is and what the answer should’ve been.
ML: What is a simplistic view of logistic regression when Y is binary?
We are just doing a linear regression on our X’s, then squishing the outcome into [0,1]
ML: In binary logistic regression, what is the formula for P(Y=1|X=x)?
For x of any dimension, it is as follows:
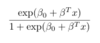
ML: What is the formula for the logit-1 function, or the inverse logit function? And what does it accomplish?
It is shown below. It squishes all real numbers x into a range of [0,1]
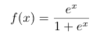
ML: In binary logistic regression, given the formula for P(Y=1|X=x), how do we choose our predicted outcome?
We of course simply choose the outcome with the higher probability.
ML: What is the final decision rule for logistic regression in its simplest form?
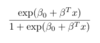
ML: What decision boundary does binary logistic regression yield?
It yields the linear separator:

ML: For binary logistic regression, how do we learn the coefficients of the model B0, B1, B2… in order to make predictions?
We estimate them using simple maximum likelihood. So we find the coefficients B0, B1, B2… that have the highest likelihood of producing the observed training data with the observed labels.
However, this optimization function using 0/1 loss is computationally hard, so we solve it iteratively using something like stochastic gradient descent.
ML: How to we extend binary logistic regression to multinomial logistic regression? So our X’s are still n-dimensional vectors, but now our Y’s are scalars in [1,K]?

ML: What issue arises when logistic regression is performed on data that is perfectly linearly separable? And how can we combat this?
The learned weights will go to infinity, and we will overfit.
We can combat this by regularizing!
ML: What does having a high number of parameters in your model do to the bias/variance tradeoff?
Having a lot of parameters, or a complex model, decreases bias (as we have more flexibility to fit whatever truly is the underlying model) but variance increases
ML: What does having a high number of features in your model do to the bias/variance tradeoff?
Having a lot of featuers decreases bias (as we are able to fit more of the possible underlying models), but variance increases due to the curse of dimensionality.
ML: In general, what kinds of changes to your model will decrease bias, but increase variance?
When you allow your algorithm the potential ability to fit a higher amount of potential underlying models, or more types of underlying models.
This of course decreases bias as you are more likely to hit the real model, but it increases variance because (I think) when you have more possible options and the same amount of data, it becomes more likely that one of them appears as the best-fitting model due simply to noise.
ML: In general, what sorts of changes to your algorithm will decrease variance, but increase bias?
Changes that make your algorithm less influenced by outliers or noise.
ML: What is a definition of model bias that is useful to think about when considering the bias/variance tradeoff?
Bias in a model is a lack of ability to fit the underlying model.
In other words, a lack of robustness with which to fit potential underlying models
ML: What is a definition of model variance that is useful to think about when considering the bias/variance tradeoff?
Model variance is its amount of susceptibility to changes due to noise.