Neuro Flashcards
(22 cards)
Normal Development - Pattern of Myelination
A full-term newborn will have myelination seen in what structures?
Myelination is most apparent on what sequence? Myelin is bright or dark on T1? How about T2?
When does myelination generally assume a mature pattern? What lobes continue to myelinate through adolescence?
- A full-term newborn will have myelination seen in several structures at birth, including posterior limb of internal capsule, middle cerebellar peduncle, ventrolateral thalami, dorsal brainstem, and the perirolandic regions.
- Myelination is most apparent on T1-weighted images. On T1-weighted images myelin is bright; on T2-weighted images myelin is dark.
- Myelination generally assumes a mature pattern by age 2 years, although the frontal lobes continue to myelinate through adolescence.
Normal Development - Overview of Cortical Development
Primitive brain form around what? What is it lined by?
Development of normal cortex is dependent on what two things?
- Primitive brain forms around the primordial ventricular system, which is lined by pluripotent stem cells called germinal matrix cells.
- Development of normal cortex is dependent on both germinal matrix cell migration and subsequent organization into the normal six-layered lamination of the cortex.
What are the CNS congenital malformations?
- Polymicrogyia
- Lissencephaly
- Schizencephaly
- Holoprosencephaly
- Gray matter heterotopia
- Agenesis/hypogenesis of corpus callosum
- Chiari I, II and Dandy-Walker in the posterior fossa
- Joubert syndrome (midbrain)
Polymicrogyria
What is it?
Caused by disturbance of what?
Etiology/Causes?
Clinical manifestations?
MR appearance?
Most common distribution?
- Polymicrogyria is a malformation of cortical development characterized by a derangement of the normal six-layered organization of the cortex, which leads to abnormally shallow and abnormally numerous sulcation.
- Polymicrogyria is thought to be caused by disturbance either late in neuronal migration or early in cortical lamellar organization.
- Polymicrogyria may be caused by in-utero infection (especially CMV), in-utero ischemia, or genetic causes.
- Clinical manifestations include developmental delay, quadriparesis, or intractable seizures.
- On MR imaging, sulci appear thick, bumpy, small, and irregular. The junction of the polymicrogyric cortex and the white matter is irregular and less well defined than normal.
- Bilateral perisylvian polymicrogyria is the most common distribution.
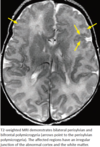
Lissencephaly
What is it characterized by?
What is it thought to be due to?
If calicifications are present, what do you think the etiology may be?
Clinical manifestation?
Subdivisions? How are they characterized?
- Lissencephaly is characterized by absent or decreased cortical convolutions causing a smooth, thickened cortical surface. Instead of the normal six-layered cortex, the cortex is arranged into four primitive layers.
- Lissencephaly is thought to be due to an arrested band of neurons, which have not completed outward migration to the superficial cortex.
- Lissencephaly may be caused by CMV, in which case calcifications are often present.
- Clinically, patients with lissencephaly almost universally have seizures.
- Lissencephaly can be subdivided into type I (classical) and type II (cobblestone). The two forms are genetically and embryologically distinct. Cobblestone lissencephaly is associated with cerebellar and ocular malformations and muscular dystrophy (Walker-Warburg syndrome).
- Type I lissencephaly is characterized by a smooth cortex and a configuration of the cerebral hemispheres that has been likened to an hour-glass or a figure-of-eight.
- Type II - cobblestone lissencephaly is characterized by a finely undulating cortex.

What is Walker-Warburg syndrome, its primary characteristics, and additional anomalies?
Walker-Warburg syndrome (WWS), sometimes known as HARDE syndrome, is an extremely rare lethal form of congenital muscular dystrophy. It is primarily characterised by:
- fetal hydrocephalus: almost always present
- neuronal migrational anomalies: agyria (cobblestone lissencephaly / lissencephaly type II)
- distinctive dorsal “kink” at the mesencephalic-pontine junction
- Dandy Walker continuum
- retinal dysplasia
- encephalocoele
- cerebellar malformations
- congenital muscular dystrophy
Additional anomalies include:
- agenesis of the corpus callosum
- microphthalmia or unilateral buphthalmos
- ocular colobomas
- microtia
- congenital cataracts
- genital anomalies in males
- cleft lip +/- palate
Grey Matter Heterotopia
What is this characterized by?
What may a clinical scenario?
Three main subtypes?
- Gray matter heterotopia is a range of disorders characterized by clusters of normal neurons in abnormal locations.
- Gray matter heterotopia may be a cause of seizures.
- Three main subtypes include periventricular nodular heterotopia, subcortical heterotopia, and marginal glioneural heterotopia.

Agenesis/Hypogenesis of the Corpus Callosum
Prevalence? Clinical manifestations?
Complete agenesis of the corpus callosum causes what secondary abnormalities in ventricular morphology?
In what progression does the corpus callosum develop? What is the most common place to find hypogenesis of the corpus callosum?
Associated anomalies??
- Agenesis of the corpus callosum is a relatively common congenital abnormality. The most common clinical manifestations of agenesis of the corpus callosum are refractile seizures and/or developmental delay.
- Complete agenesis of the corpus callosum causes secondary abnormalities in ventricular morphology. Colpocephaly (dilation of the occipital horns of the lateral ventricles) may result from decreased white matter volume posteriorly. The lateral ventricles are often parallel in orientation and widely spaced. Medial impressions on the lateral ventricles are caused by Probst bundles, which are axons that normally constitute the corpus callosum but instead pursue an aberrant course parallel to the interhemispheric fissure. The third ventricle may be enlarged and high-riding.
- The corpus callosum develops from genu (anterior) to splenium (posterior).
- Various degrees of corpus callosal hypogenesis may occur, the most common being absence of the splenium.
- Congenital corpus callosal anomalies are often associated with other midline abnormalities including a midline lipoma or interhemispheric cyst.

Holoprosencephaly
What is it?
What is it associated with? Common saying about holoprosencephaly?
Association with what type of vascular anomaly?
What are the three subtypes from more severe to least?
- Holoprosencephaly is a complex congenital malformation where the forebrain does not divide into two hemispheres.
- Holoprosencephaly is associated with midline maxillofacial anomalies including a single central incisor. It is commonly said that “the brain predicts the face.”
- Holoprosencephaly is associated with an azygos configuration of anterior cerebral artery (single ACA).
- The three subtypes (from most severe to least severe) are alobar, semilobar, and lobar.
Schizencephaly
What is it?
What is seen in up to 30% of patients with schizencephaly?
What two classical morphological classifications are there?
What other dysplasia is it associated with?
Characteristic imaging finding? Involvement is most common where?
What other entities can cause interruption or cleft in the cortex? What is one big difference between these and schizencephaly?
- Schizencephaly is a malformation characterized by a full-thickness cleft of the cerebral hemisphere lined by dysplastic gray matter (usually from polymicrogyria), forming an abnormal communication between the ventricles and the extra-axial subarachnoid space.
- Schizencephaly is associated with cortical malformations (gray matter heterotopia), with up to 30% of patients with schizencephaly also having cortical malformations.
- Schizencephaly has been morphologically classified into open-lip (walls of the cleft are entirely divided by CSF) and closed-lip (walls of the cleft are apposed or incompletely divided).
- Schizencephaly is associated with septo-optic dysplasia, which is characterized by agenesis of the septum pellucidum and optic nerve hypoplasia.
- The characteristic imaging finding of schizencephaly is a CSF-filled cortical cleft lined by gray matter. Frontal involvement is most common.
- There are several other entities that can cause an interruption or cleft in the cortex, but only a schizencephalic cleft is lined by gray matter. Other cortical clefts include:
- Porencephaly, where there is a replacement of cortex by a cystic structure.
- Encephalomalacia.
- Surgical resection cavity.

Chiari I Malformation
What is it?
Clinical manifestation? How about in babies?
Most common complication? Less common complication?
Describe the most common complication in Chiari I.
Classic criterion for diagnosis of Chiari I?
If the only finding is isolated inferior displacement of the tonsils, what term is generally preferred?
- Chiari I is inferior displacement of the cerebellar tonsils beyond the foramen magnum.
- The clinical manifestation of Chiari I is variable and ranges from occasional exertional headaches in an otherwise normal individual to severe myelopathy and brainstem compromise. In babies, Chiari I may be associated with sleep apnea and feeding problems.
- The most common complications of Chiari I are cervical syringomyelia and less commonly hydrocephalus.
- Syringomyelia, also called syringohydromyelia or syrinx , is fluid within the spinal cord. Syringomyelia most commonly represents dilation of the central canal of the spinal cord, which normally extends from the obex of the fourth ventricle to the filum terminale__. The syringomyelia associated with Chiari I is fusiform dilation of the central canal that does not typically communicate with the fourth ventricle. other forms of syringomyelia (typically associated with hydrocephalus) may communicate with the fourth ventricle via the central canal.
- Both clinical diagnosis and diagnostic imaging criteria for Chiari I are controversial. Inferior protrusion of the cerebellar tonsils is a relatively common finding in asymptomatic individuals. Additionally, the clinical symptoms of Chiari I are nonspecific and include headache and malaise, which may not always be attributable to the inferior protrusion of the tonsils.
- The classic criterion for diagnosis is herniation of either cerebellar tonsil by 5 mm, or herniation of both tonsils by 3 mm; however, associated findings allowing a more confident diagnosis include pointing of the cerebellar tonsils, crowding of the posterior fossa, and a complication such as cervical spine syringomyelia or hydrocephalus.
- If the only finding is isolated inferior displacement of the tonsils, the term “borderline tonsillar ectopia” is generally preferred.

Chiari II Malformation
What is it? What is the common feature between Chiari I and II?
What is the primary abnormality in Chiari I? What happens to the tectum?
What else is universally presented and where is it typically located?
What happens to the 4th ventricle?
Do these patients get hydrocephalus?
Associated supratentorial anomalies?
- Chiari II is a completely different disease from Chiari I; however, a common feature among all Chiari malformations is inferior displacement of the hindbrain.
- The primary abnormality in Chiari II is herniation of the vermis, cerebellar tonsils, and medulla into the foramen magnum, with resultant beaking of the tectum__.
- A myelomeningocele is universally present, typically lumbar.
- The fourth ventricle becomes elongated and inferiorly displaced. Approximately 80-90% of children with Chiari II have hydrocephalus necessitating shunt placement, due to fourth ventricular obstruction.
- Associated supratentorial anomalies include corpus callosal dysgenesis, heterotopias, and sulcation abnormalities.

Dandy-Walker Malformation
What is it?
What is the cause of this malformation? What structures are affected?
Do patients get hydrocephalus?
What happens to the tent? How about the torcula? What is the torcula?!
- The dandy Walker complex features an enlarged posterior fossa, usually associated with a large posterior fossa cyst. Dandy Walker variants are a continuum of disorders of varying severities.
- An in-utero insult to the developing fourth ventricle is thought to result in fourth ventricular oufllow obstruction, cyst-like dilation of the fourth ventricle (which communicates with a retrocerebellar cyst), and resultant inferior vermian hypoplasia.
- Most patients have hydrocephalus.
- Upwards displacement of the tentorium by the enlarged fourth ventricle and posterior fossa cyst causes torcular-lambdoid inversion. The torcular herophili is the confluence of the transverse and the straight sinuses, which is pushed superiorly, above the lambdoid suture.
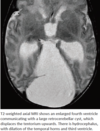
Neurofibromatosis I / von Recklinghausen Disease
What is it? What are the manifestations?
Is this a hereditary disorder?
Describe the “unidentified bright spots”
What are neurofibromas? Which parts of peripheral nerves are most commonly involved? What is the plexiform substype?
Compare and contrast neurofibromas with schwannomas. What is the “target-sign”?
Can you spare the nerve in neurofibroma resection?
- Neurofibromatosis type 1, also known as von Recklinghausen disease, is a multisystemic neurocutaneous disorder with prominent skin manifestation (e.g., café au lait spots), peripheral nerve sheath tumors (e.g., plexiform neurofibroma), CNS malignancies (e.g., optic nerve glioma), and bony abnormalities (e.g., sphenoid wing dysplasia).
- NF1 is autosomal dominant in 50% of cases and occurs sporadically in 50%, caused by a defect in chromosome 17.
- In children, “bright spots” or “unidentified bright objects” are almost universally seen on T2-weighted images, thought to be caused by myelin vacuolization. These bright spots are seen most commonly in children ages 4-12 and become less prominent in adults.
- Neurofibromas are WHO grade I nerve sheath tumors. The cutaneous and subcutaneous nerves are more commonly involved than the more proximal peripheral nerves. A plexiform subtype is more aggressive and consists of a network of fusiform-shaped masses, with malignant degeneration reported in approximately 5%.
- Like schwannomas, neurofibromas are likely of Schwann cell origin. The target sign can be seen with either neurofibromas or schwannomas, and reflects central T2 hypointensity thought to be due to a fibrocollagenous core. The target sign is suggestive of benignity.
- In contrast to schwannomas, neurofibromas are not encapsulated and involve the entire cross-sectional area of the nerve. If a neurofibroma is resected, the parent nerve must, therefore, be sacrificed.

What are the NF1 associated brain neoplasms?
- Optic nerve glioma (50% of these tumors are associated with NF1).
- Juvenile pilocytic astrocytoma.
- Brainstem glioma.
What are the NF1 associated bone manifestations?
- Sphenoid wing dysplasia, which may produce pulsatile enophthalmos or exophthalmos.
- Posterior vertebral body scalloping.
- Rib notching (twisted ribbon ribs) due to erosion from neurofibromas of the intercostal nerves.
- Focal gigantism.
- Cervical kyphoscoliosis, with a characteristic acute angle.
- Neural foraminal enlargement, either due to bony dysplasia or a neurofibroma.
- Tibial bowing.
What is the eye manifestation of NF1?
- Eye manifestations of NF1 include hamartomas of the iris, known as Lisch nodules.
What are the NF1 associated extra-cranial neoplasms?
- Wilms tumor.
- Rhabdomyosarcoma.
- Angiomyolipoma (renal).
- Leiomyosarcoma.
Neurofibromatosis 2
What is it? Contrast to NF1
What is this disorder characterized by? Include the mnemonic!
What is the classical clinical presentation of NF2?
Presence of what imaging finding is diagnostic of NF2?
- Neurofibromatosis type 2 - NF2 is an autosomal dominant neurocutaneous disorder completely unrelated to NF1. Despite the name, neurofibromas are not a component of NF2. NF2 is caused by a defect on chromosome 22 and is approximately ten times less common than NF1.
- NF2 is characterized by the MISME mnemonic: Multiple Inherited Schwannomas, Meningiomas, and Ependymomas.
- The typical clinical presentation of NF2 is hearing loss caused by bilateral vestibular schwannomas. The presence of bilateral vestibular schwannomas is diagnostic of NF2.

Sturge Weber Syndrome
What is it characterized by?
Where does the “stain” typically involve? Corresponds to which nerve?
What kind of disorder is this? What does this result in?
The underlying mechanism of this disorder results in what?
Imaging appearance on CT? On contrast-enhanced MR?
- Sturge Weber is a neurocutaneous disorder characterized by facial port-wine stain (capillary malformation), ocular abnormalities, and failure of normal cortical venous development.
- The port-wine stain typically involves the forehead and upper eyelid, corresponding to the region innervated by the ophthalmic branch of the trigeminal nerve (V1).
- Sturge Weber is a vascular disorder, thought to be caused by the failure of regression of the primitive embryologic cephalic venous plexus. This developmental anomaly results in the formation of leptomeningeal venous angiomatosis, which is a vascular malformation characterized by dilated capillaries and venules.
- The underlying vascular anomaly ultimately leads to chronic ischemia, cortical atrophy, and cortical calcification.
- Clinically, patients may have some degree of developmental delay and seizures; however, up to 50% of patients may have normal intelligence. Seizures and developmental delay tend to occur together.
- Characteristic imaging findings are cortical atrophy and subcortical parenchymal calcifications, which are best seen on CT. Contrast-enhanced MRI demonstrates pial enhancement in regions affected by the leptomeningeal venous angiomatosis. The choroid plexi are typically enlarged from compensatory hypertrophy thought to be related to increased flow.

Tuberous Sclerosis
What is it?
Classical clinical tried?
Clinical manifestations?
Neuroimaging appearance?
15% of patients develop what CNS tumor? Appearance?
Included extracranial manifestations?
- Tuberous sclerosis is a hamartomatous disorder affecting several organ systems, with multiple skin manifestations.
- The classical clinical triad of adenoma sebaceum (nodular rash originating in the nasolabial folds), seizures, and mental retardation is not always present.
- Clinically, most patients have epilepsy, neurocognitive dysfunction, and pervasive developmental disorders.
- Neuroimaging features multiple T2 hyperintense white matter cortical or subcortical tubers (hamartomas) and subependymal nodules.
- 15% of patients develop subependymal giant cell astrocytoma (SEGA), which may appear on imaging as a newly enhancing or enlarging subependymal nodule.
- Extracranial manifestations include:
- Renal: multiple angiomyolipomas.
- Cardiac: Rhabdomyoma.
- Lung: Lymphangioleiomyomatosis.

Joubert and Related Syndrome
What characteristic configuration is seen in this disease?
Clinical presentation?
Primary abnormality in this disease?
This is often associated with what other anomalies?
Dysplastic tissue is often present where?
Contrast to Dandy-Walker
- A “molar tooth” configuration of the midbrain can be seen in several diseases, including Joubert syndrome and related disorders (JSRD).
- JSRD clinically present as a hypotonic child with developmental delay and ataxia.
- These disorders are often associated with ocular anomalies.
- The primary abnormality of Joubert syndrome is thought to be aplasia or hypoplasia of the cerebellar vermis. Dysplastic cerebellar tissue is often present.
- Unlike Dandy-Walker complex, hydrocephalus and a large posterior fossa cyst are uncommon.

