Unit 2 - Cell Cycle Flashcards
(89 cards)
key tasks for proliferating cells
- replicate entire genome (ONCE) - fertilised egg → entire organism, cellular regeneration, in response to injury
- separate duplicated chromosomes equally into daughter cells (UNLIKE stem and germ cells)
- co-ordinate cell growth and proliferation by ensuring cells have enough energy and metabolites before entering into the processes
key events of the cell cycle

Gap phase G1
cell growth and regulatory events (checkpoints)
synthesis phase (S)
DNA replication occurs
gap phase 2 G2
cell growth and regulatory events (checkpoints)
M phase - mitosis
chromosomes are segregated and cells divide
when may cells exit cycle into G0
if terminally differentiated, senescent or if inhibitory signals are received
cell cycle timing
varies between cells depending on function and origin
Our genetic material is very large and will take a while to duplicate accurately, and segregate it between daughter cells
enzymes are conserved

DNA polymerases
highly conserved enzymes
require a 3’-OH group for activity and so require a primer
needed to extend DNA strands in a 5’ → 3’ direction
replicative polymerases = Pol δ and Pol ε
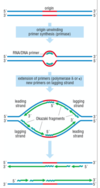
DNA replication overview
DNA is unwound and RNA primer molecules bound are synthesised by primase
primers are then extended by replicative polymerases (δ or ε) in a 5’ → 3’ direction
⇒ lagging strand is discontinuously synthesised
also leads to ‘end replication problem’ which is solved by telomerase
primase function
extend chains of DNA against the template provided by the pre-existing chromosome
Okazaki fragments
discontinuous segments being synthesised from RNA primer
telomerase function
allow intact replication of RNA ends
arises from the need of our polymerases to have our primer structure
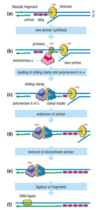
steps in DNA synthesis - lagging strand
- helicase unwinds DNA, RPA loads - Pol α-primase synthesises a short primer
- Pol α displaced and Pol δ/Pol ε loaded
- Pol δ/Pol ε extend the primer
- downstream primer is removed (nuclease)
- Okazaki fragments are ligated
SIMILAR PROCESS OCCURS ON LEADING STRAND, ensuring both strands of DNA molecule are going to be duplicated completely
Pol α-primase function
- Synthesise a short RNA primer against the template provided by the original DNA strand
- Allow replicate of polymerases (Pol delta or epsilon) to extend in 5’ to 3’ direction from the 3’-OH in a process that will give rise to the new DNA strand
- Primer is then removed by nuclease activty
- Individual extended primers, now the actual DNA sequences, are ligated together again to give a continuous new DNA strand that will base pair with the original DNA strand and will be complementary to it due to extend of polymerase
M phase overview
chromosomes condense and attach to microtubules from mitotic spindle
all chromosomes must be attached before sister chromatids can separate
each daughter cell receives 1 set of chromosomes
chromosome segregation is irreversible so this process is highly-regulated

what happens when all chromatids have attached to the poles of mitotic spindle
the sister chromatids - duplicated pairs of chromosomes - will separate from one another
(held together after replication but prior to separation during mitosis)
6 phases of mitosis
prophase
prometaphase
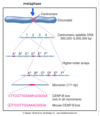
metaphase
anaphase
telophase
cytokinesis
prophase
chromosomes begin to condense
requires condensin and DNA topoisomerase II
the (duplicated) centrosomes separate
histones undergo mitosis-specific modifications
Lose their diffused localised volume and begin to adopt a more tightly packed conformation that is mechanically necessary for them to migrate to opposite poles later in mitosis
intact nuclear envelope
what makes up chromatin
histones
prometaphase
microtubules from opposite spindle poles (centrosomes) bind chromosomes at kinetochores (waist) to initiate bipolar orientation
nuclear envelope breakdown occurs - can now spread out into the cell
metaphase
all chromosomes have made bipolar attachemnts to spindle poles
chromosomes align at metaphase plate

what is the key tightly-regulated step in mitosis
between metaphase and anaphase
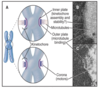
anaphase
chromatids separate and move toward the opposite spindle poles - poles separate
nuclear envelope reassembly commences - reassembles around the chromosomes as they move apart