What is Sanger sequencing?
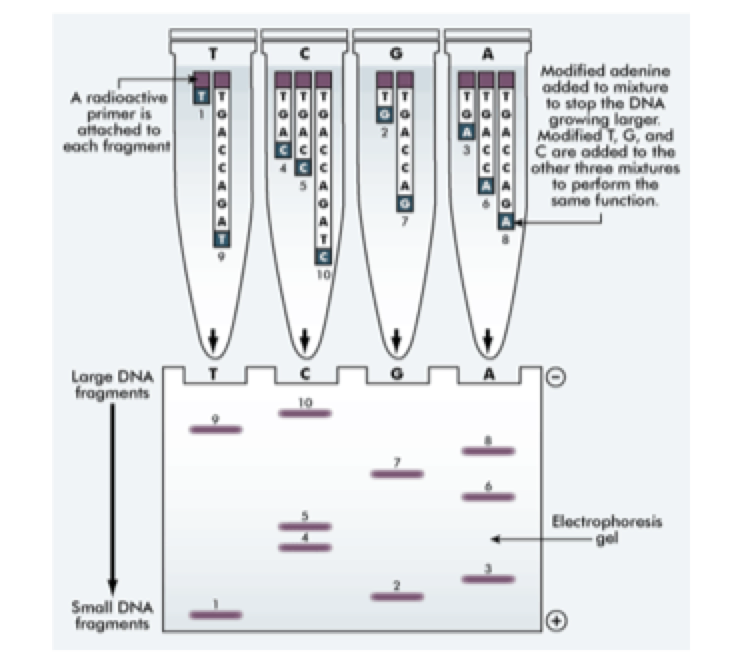
Method to determine the DNA sequence –> exploits the fact that dideoxynucleotides terminate replication –> Hence if we add a specific dideoxynucleotide (I.e. ddATP or ddCTP or ddTTP or ddGTP) it will bind to the complementary base (A-T and C-G) and terminate replication –> the point of termination corresponds to the location of the complementary base.
Hence, when we run an agarose gel we will get DNA fragments of different lengths –> each corresponding to the location of a particular base.
What does a tube for Sanger sequencing contain?
DNA replication is performed in four separate tubes, each containing:
- Single-stranded DNA to be sequenced
- DNA polymerase
- Primers
- The four dNTPs (dATP, dCTP, dTTP and dGTP)
- Small amount of one of the four 2’,3’-dideoxy analog (ddATP or ddCTP or ddTTP or ddGTP)
Note - Either the primers or the dNTPs are radiolabelled with 32P or fluorescent labels –> allows us to visualize it on an agarose gel.
Outline the procedure of Sanger Sequencing.
- A short piece of DNA called a primer is added –> the primer will bind specifically to a DNA sequence –> serves as a starting point for DNA replication.
- Primers are elongated using DNA polymerase.
If this were all, the reaction would copy a new chain until it stopped. However, this is not the case as we have dideoxynucleotides.
Dideoxynucleotides –> Lack a -OH on both the 2I and 3I carbon –> This means that when a dideoxy base is incorporated into a DNA molecule, the chain stops or terminates –> as the phosphodiester backbone can not be extended.
- At any position, either a normal base will be added, so the chain can continue to grow, or a dideoxy base will be added, so the chain terminates –> After many cycles of replication –> we get many DNA chains of different length –> each corresponding to a particular base.
- The base sequence can then be read off using the agarose gel.
How has Sanger sequencing been improved?
Modern DNA sequencing.
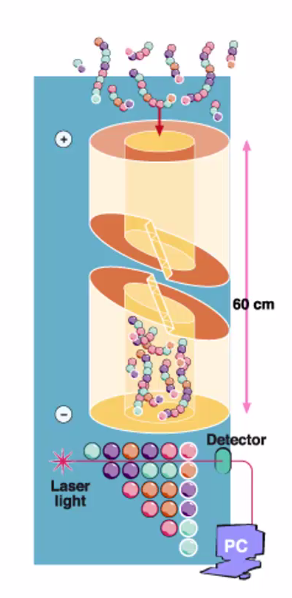
The DNA sequences are separated by size on a gel-filled capillary tube –> DNA added to one end of the gel and a charge is applied to the tube and the DNA moves through according to size, smallest first –> electrophoresis.
Reading the sequence is done by illuminating the DNA, just before it emerges, with a laser to detect the ‘coloured’ tag on the dideoxy base (each dideoxy-base has a coloured tag) at the end of the DNA copy –> The colour of the emitted fluorescence is read by the detector and a base is assigned.
The result is stored and assessed by software designed to test how reliable the base assignment is.
Note –> Peaks created are perfect –> Hence, we need a score in order to gauge validity.
What is the Phred quality score (Q-Value) used for in the modern sequencing?
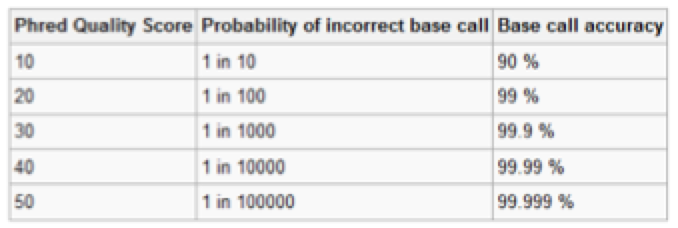
Each nucleotide read is assigned a quality score based on how confident the read prediction is –> Generally called the Q value.
The most commonly used method is to only count the bases with a quality score of 20 and above (99% accuracy)
All sequencing technologies (Illumina etc) provide a quality score for each nucleotide in their sequences
Examples of next gen sequencing methodologies?
- Illumina
- Lifetechnologies –> Ion Torrent
- Pacificbiosciences
- Nanoporetech
Outline how Illumina performs DNA sequencing?
Illumina –> most widely used.
Difference between Sanger and Illumina –> Sanger sequences a specific DNA sequences whereas with Illumina you get the entire human genome and break it apart into small DNA fragments.
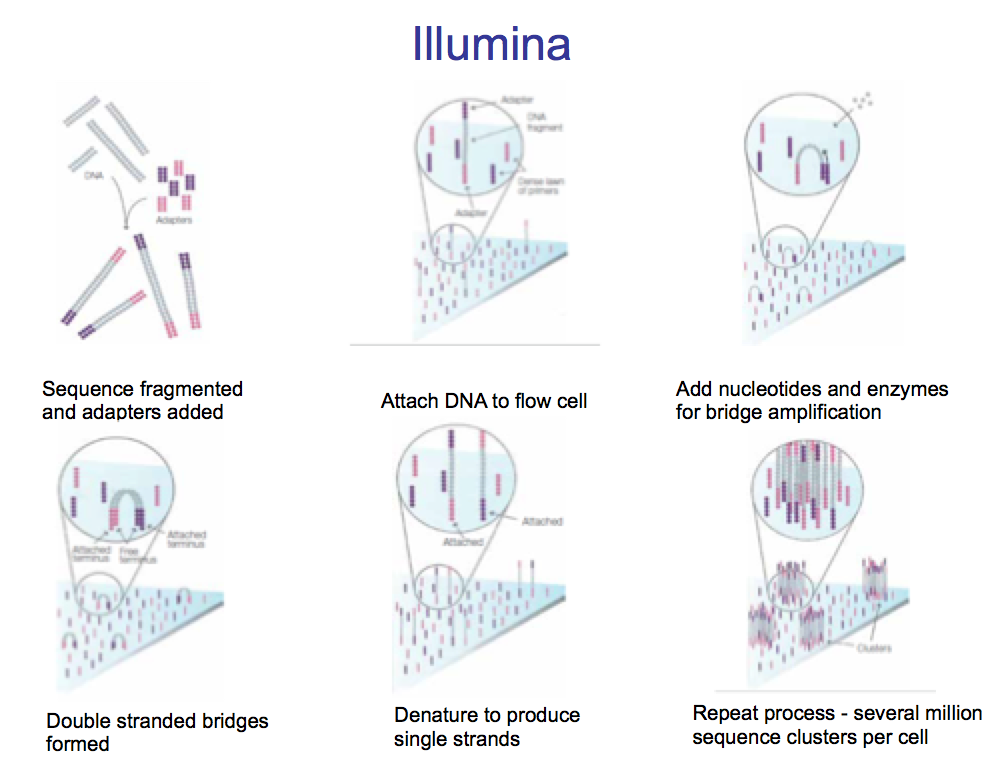
- Fragment the entire genome into small DNA fragments.
- DNA is made single-stranded
- Add Adaptors to the end of all the DNA fragments
- Add the DNA to a flow cell –> the flow cell has probes fixed to it which match the adaptors.
- As each DNA fragment has two adaptors it bridges across to bind the other adaptor to another probe.
- Add nucleotides and polymerases –> builds double-stranded sequence
- Denature the dsDNA and repeat process to produce several million sequence clusters.
Now that we have amplified the DNA we can start sequencing it.
Outline how we are able to read the DNA sequence using Illumina.
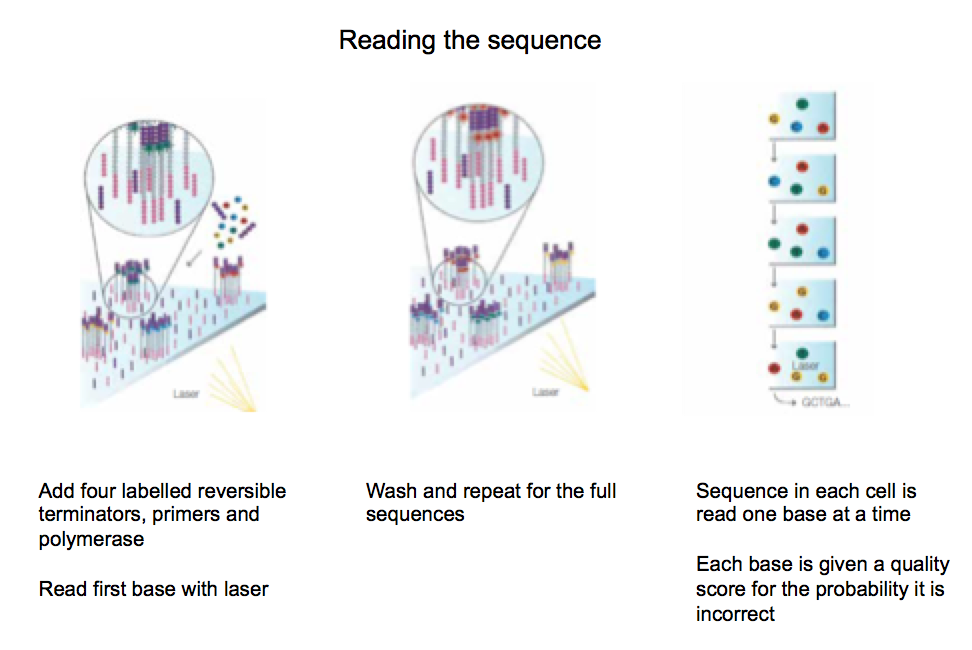
Add four labelled reversible terminators, primers and polymerase.
When the reversible terminator binds –> no other nucleotides can bind –> but it is reversible so it can be removed –> allowing for another nucleotide to bind and to be removed.
This process repeats itself –> allows us to read one base at a time.
So the process is as follows…
First reversible terminator binds –> Read the sequence with a laser –> wash to remove base –> repeat.
Note –> each nucleotide is labelled with a different colour –> allowing the laser to distinguish between them.
Once again, each base is given a quality score for the probability it is incorrect.
Problem with Illumina?
Can only read relatively short sequences at a time –> sometime only 150-200 nucleotides at a time.
Problematic for genome assembly because you need to find the position of each sequence relative to each other (examining overlap)
Longer reads would be more useful.
What is Pac Bio?
Produces the longest reads – approx 20 000
It has a high error rate of approx 87% –> however, errors are random so compensated by multiple reads and creating consensus –> Consensus method creates 99.999% accuracy.
Basically, when you sequence a genome multiple times –> the errors are random (all over the place) –> we can align the sequences and get an overall consensus (chance of an error in the same position in multiple reads is very unlikely) –> to generate an accurate read.
Would not be possible if errors were focussed on particular areas or sequence patterns
Why can PacBio produce longer reads than Illumina?
- Polymerase used by Illumina falls off DNA quite easily –> struggle to keep DNA poly on.
However…
PacBio uses polymerase from a deep-sea sponge –> adapt in order to allow biological functions to occur at extreme pressures –> hence, the polymerase has to bind tightly to the DNA.
Thus, being an advantage for gene sequencing –> allows us to create longer reads.
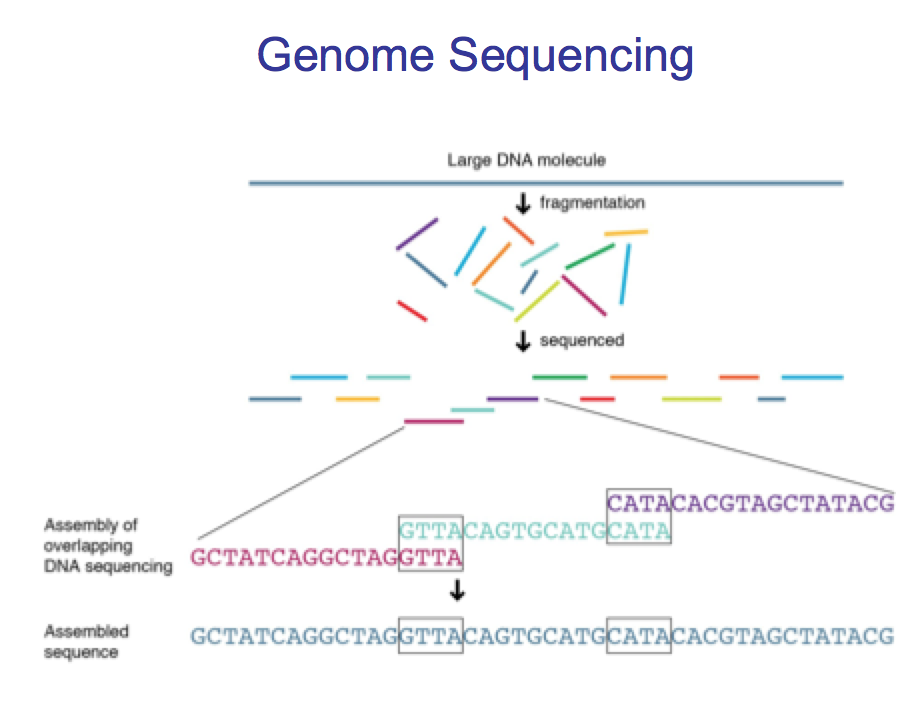
How do we go about sequencing a genome?
After sequencing a genome (sanger or next-gen sequencing) we end up with all these fragments of DNA that have been sequenced which will need to put together –> like a puzzle –> Known as de novo assembly
Note –> this is not as problematic now as we have already sequenced the human genome which we can use as a reference.
Method - Shotgun sequencing
- The genome sequence is shredded into pieces and inserted into plasmids
- We sequence each fragment from both ends –> get reads –> this process is repeated for all fragments of the genome
- The sequence is then assembled de novo or against a reference for comparison
The basic idea behind De Novo sequencing?
Get genome –> convert it into a lot of fragments –> sequence –> examine for overlaps –> if there is sufficient overlap we know that those two fragments are adjacent to each other –> over time build up the genome.
In principal seems easy but in reality not because…
We have millions of fragments + we have a lot of repetitive sequences.
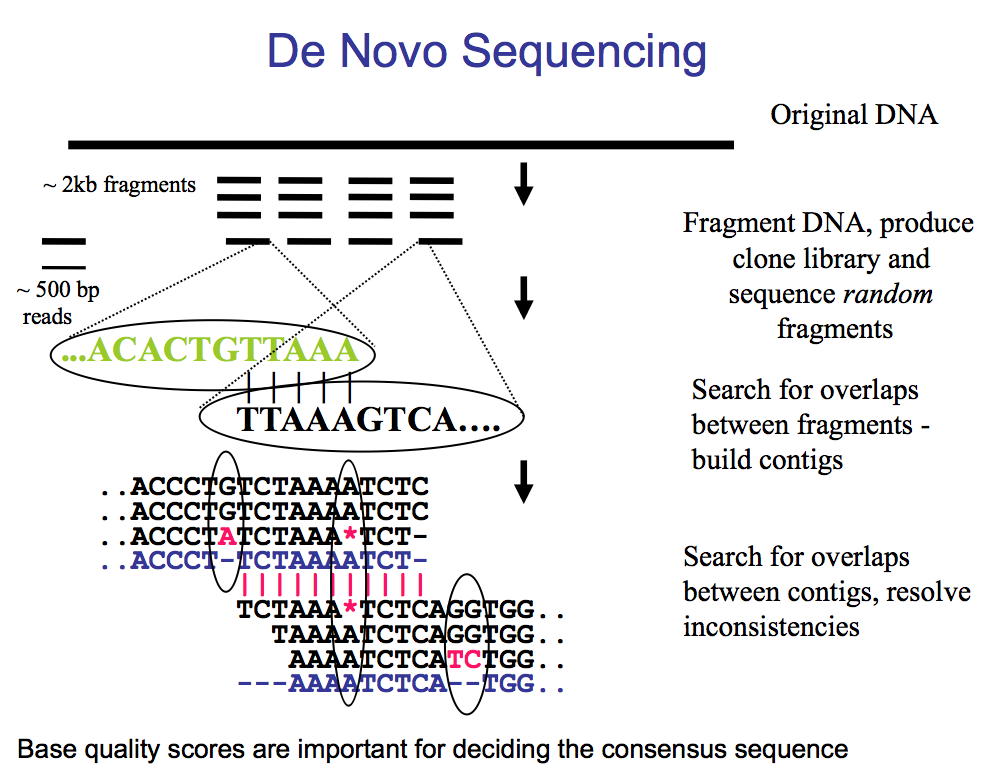
Outline De novo sequencing.
De novo sequencing
We get fragments and search for overlaps to build up a consensus.
But at one point you will reach an error –> this is where we use the quality score in order to gauge whether we should consider this difference or not –> quality score low we disregard error.
What does the ‘Depth of Coverage’ refer to?
Sequencing errors are eliminated by the depth of coverage of overlapping sequence fragments.
For the Human Genome Project, most of the genome was sequenced at 12X or greater coverage.
This means that each base was present in 12 reads on average –> by increasing the number of times we sequencing and checking the overlap –> the higher probability that overlap is correct.
However…
Even with 12x coverage approximately 1% of the genome not accurately assembled
General rule of thumb - More complex genomes need more depth of coverage.

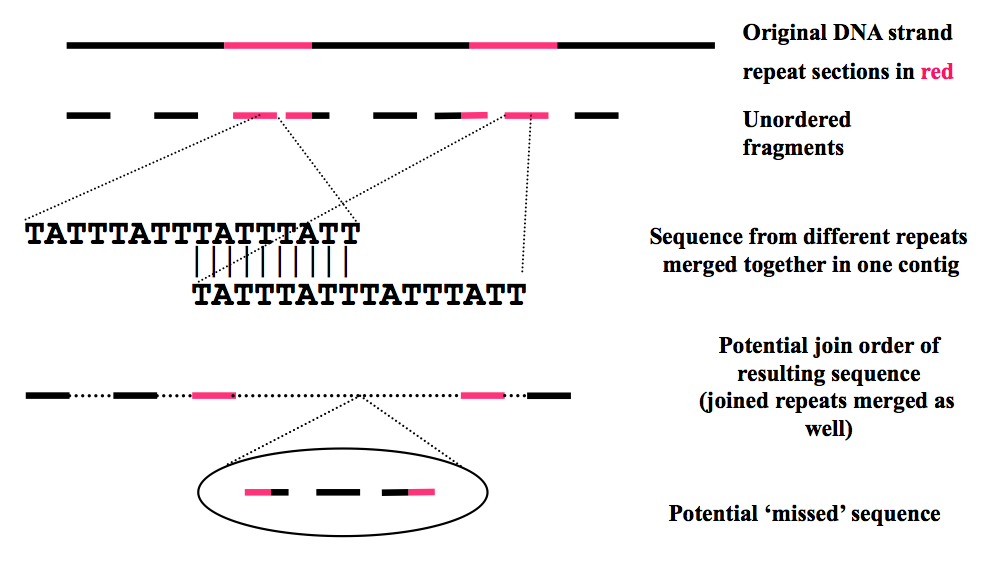
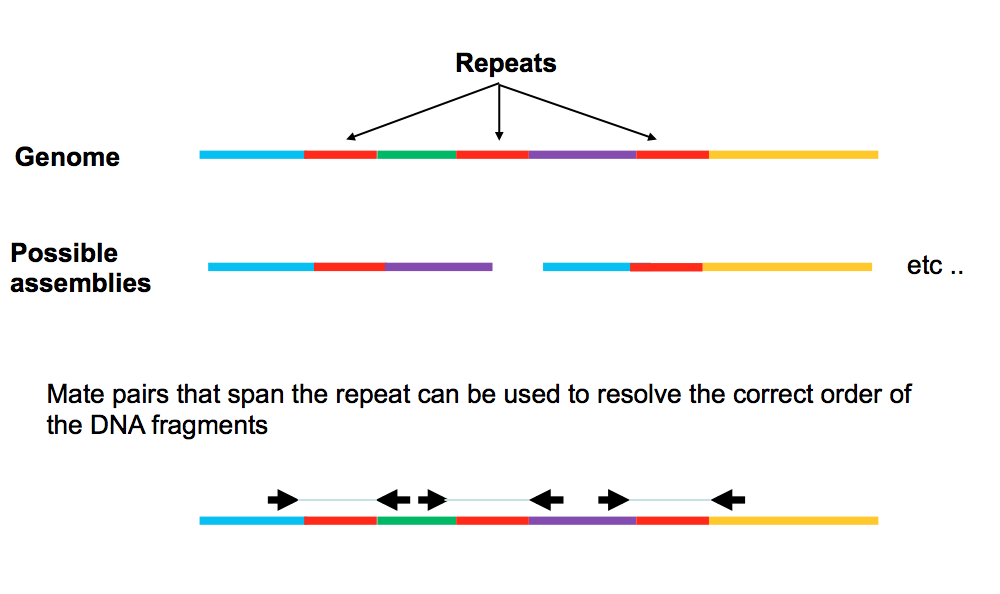
What is one of the challenges of De novo sequencing?
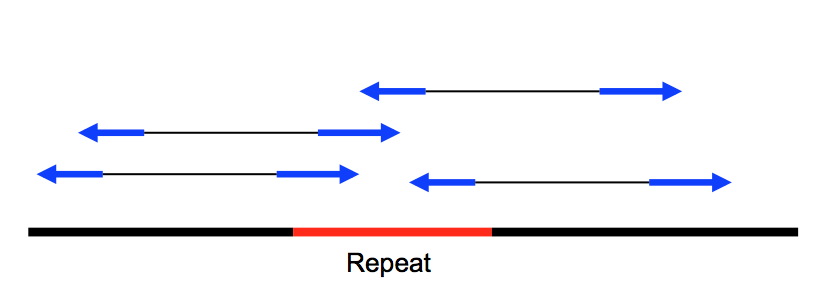
Highly repetitive DNA –> you don’t know whether this repeat fragment is from one part of the genome or the other.
If you don’t assembly correctly –> you can lose a large portion of the sequenced DNA between the two repeats (as shown in the picture).
How can paired-end reads be used to deal with repetitive DNA?
Paired End Reads
Paired-end reads are sequences from both ends of a DNA fragment. We know the paired ends because we sequence from both sides of each fragment –> so we know what’s on each end but not necessarily what’s in the middle.
If the fragment is 700bp and the reads 100bp they provide 3 pieces of information:
- the tag 1 sequence
- the tag 2 sequence
- that they were 500bp apart in your genome (distance)
This gives you the ability to map to a reference (or denovo) using that distance information. It helps resolve structural rearrangements (insertions, deletions, inversions), as well as helping to assemble across repetitive regions.
How exactly are Pair reads used?
Since we know the Paired-end repeat sequence and the distance between them we can start building up the genome –> as we know that they must be a specific distance apart.
If one read is unmappable because it falls in a very repetitive region but the other end is unique, you can use that distance information to map both reads.
Basically, we know one end of the which acts as anchors –> whereas, the other end falls in a repetitive region –> normally we wouldn’t be able to distinguish where this repetitive sequence is from but since we know that it must be a certain distance from the anchor we know its position.
Normally works for short repeats not longer repeats.
What do we use for large repeats?
Use this when the repeat is larger (Few Kb) than the distance we can read.
Use Mate Pairs –> Same approach as paired ends.
We sequence on either end of a very large fragment -> we know that each end of the sequence goes together + we know the distance apart.
Allow us to bridge across large regions and then bridge across these two ends using paired-end reads.
How can paired-end reads be used to understand cancer genomes?
Paired-end reads have a lot of DNA rearrangements.
Sequence cancer genome and reference it back to the reference genome.
What we want to find out is how the cancer genome varies from the reference.
We get paired-end reads which are X distance apart –> when mapping it back if it is less than X distance this means that we have an insertion in the cancer genome. Likewise, if the reference distance is more than X distance than we have had a deletion in the cancer genome.
We can also identify inversions (the sequence is inverted) or translocation (movement of sections to a different chromosome)
What is scaffolding?
When sequencing we create contigs –> Contiguous sequence where base order is known —> Assembled from sequence reads.
But we will always get gaps –> use mate pairs to figure out the order of contigs.
Note - Approx length of fragments are known so we number of base pairs between contigs.
Hence, a Scaffold is a…
Genome sequence reconstructed from contigs and gaps.
To fill the gap we need to manually sequence using chromosome walking.
What are some assembly limitations?
Denovo assembly of complex genomes is still problematic –> Even greater problem for next gen sequencers with small read lengths –> Will improve with longer reads and 3rd gen sequencers.
Examples of difficult genomes
Entamoeba hystolytica
- Very AT rich genome
- Ploidy unknown
- Over 1500 contigs
- Genome size approx 20Mb
Blumeria graminis
- Repeat rich
- Approx 7000 contigs
- Genome size approx 120Mb
Benefits of Next-Gen sequencing?
Next generation sequencers can be used for:
- Genome sequencing/re-sequencing –> looking for variants
- Targeted resequencing
- SNP detection
- Transcriptome sequencing for expression analysis, and splice variant detection (RNA-Seq)
- Protein-DNA/RNA interactions (ChIP-Seq) –> protein binding sites
- DNA Methylation (MeDIP-Seq) –> methylation patterns.
- Seq –> means next-gen sequencing.
What is genome annotation?
Going from the basic DNA sequence –> to the fully annotated version –> introns, exons, transcription factor binding sites, repeats, etc.


















-
DNA structure - Lecture53
-
DNA structure - Book42
-
Kenji - Lectures85
-
Kenji - Book27
-
DNA replication - Lecture45
-
DNA replication - Book45
-
Transcription/Translation - Lecture88
-
Transcription/Translation - Book95
-
Gene Cloning - Lecture70
-
Gene cloning - Book25
-
Eukaryotic Genetics - Lecture165
-
Bacterial Genetics - Lecture99
-
Genomics - Lecture64
-
Gene Regulation60
